FDA Approves Monovisc Injection for Knee Pain

Sodium Hyaluronate
9067-32-7 (sodium salt)
| MF: | C14H22NNaO11 |
| MW: | 403.31 |

26 feb 2014
Anika Therapeutics Inc. announced it has received marketing approval for Monovisc from the U.S. Food and Drug Administration (FDA). Monovisc is a single injection supplement to synovial fluid of the osteoarthritic joint, used to treat pain and improve joint mobility in patients suffering from osteoarthritis (OA) of the knee.
Monovisc is the first FDA-approved, single-injection product with HA from a non-animal source. It is comprised of a sterile, clear, biocompatible, resorbable, viscoelastic fluid composed of partially cross-linked sodium hyaluronate (NaHA) in phosphate buffered saline.
read all at

Sodium hyaluronate is the sodium salt of hyaluronic acid, a glycosaminoglycan found in various connective, epithelial, and neural tissues. Sodium hyaluronate, a long-chain polymer containing repeating disaccharide units of Na-glucuronate-N-acetylglucosamine, occurs naturally on the corneal endothelium, bound to specific receptors for which it has a high affinity. The polyanionic form, commonly referred to as hyaluronan, is a visco-elasticpolymer normally found in the aqueous and vitreous humour. As a pharmaceutical, the uses of sodium hyaluronate include:
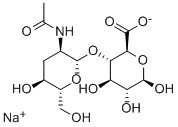
sodium hyaluronate
Sodium hyaluronate for intra-articular injection (brand names: Euflexxa, Hyalgan, Supartz, Gel-One) is used to treat knee pain in patients withosteoarthritis who have not received relief from other treatments. It is very similar to the lubricating fluid that occurs naturally in the articular capsule of the knee joint. Once injected into the joint capsule, it acts as both a shock absorber and a lubricant for the joint.[1]

Sodium hyaluronate for intraocular viscoelastic injection (brand names: Healon, Provisc, Viscoat) is used as a surgical aid in variety of surgical procedures performed on the eyeball including cataract extraction (intra- and extracapsular), intraocular lens implantation, corneal transplant,glaucoma filtration, and retina attachment surgery. In surgical procedures in the anterior segment of eyeball, instillation of sodium hyaluronate serves to maintain a deep anterior chamber during surgery, allowing for efficient manipulation with less trauma to the corneal endothelium and other surrounding tissues. Its viscoelasticity also helps to push back the vitreous face and prevent formation of a postoperative flat chamber. In posterior segment surgery, sodium hyaluronate serves as a surgical aid to gently separate, maneuver, and hold tissues. It creates a clear field of vision, facilitating intra-operative and post-operative inspection of the retina and photocoagulation.[2]
Sodium hyaluronate is used as a viscosupplement, administered through a series of injections into the knee, increasing the viscosity of the synovial fluid, which helps lubricate, cushion and reduce pain in the joint.[3] It is generally used as a last resort before surgery[4] and provides symptomatic relief, by recovering the viscoelasticity of the articular fluid, and by stimulating new production from synovial fluid.[5] Use of sodium hyaluronate may reduce the need for joint replacement.[6] Injections appear to increase in effectiveness over the course of four weeks, reaching a peak at eight weeks and retaining some effectiveness at six months, with greater benefit for osteoarthritis than oral analgesics.[7] It may also be effective when used with other joints.[8]
Sodium hyaluronate may also be used in plastic surgery to reduce wrinkles on the face or as a filler in other parts of the body.[9] It may be used in ophthalmology to assist in the extraction ofcataracts, the implantation of intraocular lenses, corneal transplants, glaucoma filtration, retinal attachment and in the treatment of dry eyes.[10]
Sodium hyaluronate is also used to coat the bladder lining in treating interstitial cystitis.
 hyaluronan
hyaluronan
cas 9004-61-9
Sodium hyaluronate functions as a tissue lubricant and is thought to play an important role in modulating the interactions between adjacent tissues. Sodium hyaluronate is a polysaccharide which is distributed widely in the extracellular matrix of connective tissue in man. It forms a viscoelastic solution in water which makes it suitable for aqueous and vitreous humor in ophthalmic surgery. Mechanical protection for tissues (iris, retina) and cell layers (corneal, endothelium, and epithelium) are provided by the high viscosity of the solution. Elasticity of the solution assists in absorbing mechanical stress and providing a protective buffer for tissues. This viscoelasticity enables maintenance of a deep chamber during surgical manipulation since the solution does not flow out of the open anterior chamber. In facilitating wound healing, it is thought that it acts as a protective transport vehicle, taking peptide growth factors and other structural proteins to a site of action. It is then enzymatically degraded and active proteins are released to promote tissue repair.[11] Sodium hyaluronate is being used intra-articularly to treat osteoarthritis.
Sodium hyaluronate is an ophthalmic agent with viscoelastic properties that is used in joints to supplement synovial fluid.
Sodium hyaluronate is absorbed and diffuses slowly out of the injection site. It is eliminated via the canal of Schlemm.
Sodium hyaluronate hyaluronan started to be in use to treat osteoarthritis of the knee in year 1986 with the product Hyalart/Hyalgan by Fidia of Italy, in intra-articular injections.
Sodium Hyaluronate
Brand names of Sodium hyaluronate in Market include (alphabetically):
- AMO Vitrax (ocular)
- AMVISIC Plus (ocular)
- CYSTISTAR, Healon (ocular)
- EYEFILL (ocular)
- HYLO-COMOD (Eye Drop)
- OLIXIA Pure (Eye Drop)
- EUFLEXXA, Bio Technology General (Israel)-Meditrina SA (Rx articular), Molecular weight: 2,400,000-3,600,000 Daltons
- GONILERT/Verisfield (UK) (Rx/articular). Molecular weight:1,800,000-2,000,000 Daltons
- HYALGAN/HYALART- Fidia (Italy)(Medical Device/Rx articular)
- MONOVISC- Anika (USA)(MedicalDevice/articular)
- OSTENIL- TRB Chemedica (Switzerland)(articular injection) [1]
- RECOSYN- Merckle Recordati (Germany) Recosyn info leaflet
- SYNOCROM- Croma Pharma (Austria) (articular injection) . Molecular weight:1,600,000 Daltons
- VISCURE- Cube (UK)(Rx/articular), Molecular weight:1,800,000-2,000,000 Daltons
- VISMED- TRB Chemedica (Switzerland)(eye drop)[2]
- YARDEL- Libytec (Impfstoffwerk Dessau-Tornau/Germany,(Rx/articular), Molecular weight:1,800,000-2,000,000 Daltons
Hyaluronic acid (HA) is a glycosaminoglycan which is present in the hyaline cartilage, synovial joint fluid and skin tissues. More particularly, HA is a linear glycosaminoglycan formed by a mixture of chains of different length constituted by the repetition of a regular disaccharide formed by a glucuronic acid unit and a N- acetyl-glucosamine unit linked beta 1-4. Disaccharides are linked beta 1-3 with an average molecular weight up to 6 Md (6×106 Da). Therefore, each chain in said mixture of chains shows the same repetitive sequence of formula (A)

the corresponding cation generally being hydrogen (hyaluronic acid) or sodium (sodium hyaluronate).
In the tissues, the function of hyaluronic acid is mainly to maintain the structural density allowing in the same time the biochemical actions of the natural products in the specific body districts. In fluids like synovia the action of HA is to keep the right viscosity by a lubricant action. To exert these actions, HA needs to be fully biocompatible including a right metabolic balance. Natural HA is continuously degraded and synthesized by the body enzymes. This homeostasis is deviated when pathological situations occur, therefore increases in the HA catabolism can results in wide range of effects from a severe pathology to simple tissue modifications. The application of HA, as sodium hyaluronate, as filler in cosmetic or in viscoelastic replacement in synovitis, requires that the employed HA polymer has enhanced viscoelastic properties. This rheology has to be balanced with an efficient capability to make the production of the injectable product.
The industrial hyaluronic acid is obtained by extraction from animal tissues or by microorganism fermentation and is commonly available as sodium hyaluronate. Concerning molecular weight, it is generally recognized that low molecular weight HA is a mixture of chains having a mean molecular weight below 250 Kd (2.5×105Da). HA is used, generally as sodium hyaluronate, in many applications in cosmetics, ophthalmology, rheumatology and tissues engineering. In particular HA with a mean molecular weight above 1 Md is used as viscosupplement in joint arthrosis or in wrinkle management. The high molecular weight is required to supplement the synovial fluid or to fill skin connective dead spaces thanks to the viscosity of the resulting solution.
Many medicaments based on the above technology are currently available on the market. They have a high biocompatibility but they are subjected to a rather rapid degradation by the body enzymes, in particular by hyaluronidase, with the consequence of a short half-life.
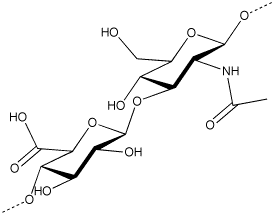 sodium hyaluronate
sodium hyaluronateReferences
- “Hyaluronate sodium: Indications, Side Effects, Warnings” (Web). Drugs.com. Drugs.com. 5 February 2014. Retrieved 25 February 2014.
- “Healon (Sodium Hyaluronate)” [package insert]. (2002). Kalamazo, Michigan: Pharmacia Corporation. (Web). RxList. (Updated 8 December 2004). RxList, Inc. Retrieved 25 February 2014.
- Puhl, W.; Scharf, P. (1997). “Intra-articular hyaluronan treatment for osteoarthritis”. Annals of the rheumatic diseases 56 (7): 441. doi:10.1136/ard.56.7.441. PMC 1752402.PMID 9486013. edit
- Karlsson, J.; Sjögren, L. S.; Lohmander, L. S. (2002). “Comparison of two hyaluronan drugs and placebo in patients with knee osteoarthritis. A controlled, randomized, double-blind, parallel-design multicentre study”. Rheumatology (Oxford, England) 41 (11): 1240–1248.PMID 12421996. edit
- Jubb, R. W.; Piva, S.; Beinat, L.; Dacre, J.; Gishen, P. (2003). “A one-year, randomised, placebo (saline) controlled clinical trial of 500-730 kDa sodium hyaluronate (Hyalgan) on the radiological change in osteoarthritis of the knee”. International journal of clinical practice 57 (6): 467–474. PMID 12918884. edit
- Kotz, R.; Kolarz, G. (1999). “Intra-articular hyaluronic acid: Duration of effect and results of repeated treatment cycles”. American journal of orthopedics (Belle Mead, N.J.) 28 (11 Suppl): 5–7. PMID 10587245. edit
- Bannuru, R. R.; Natov, N. S.; Dasi, U. R.; Schmid, C. H.; McAlindon, T. E. (2011). “Therapeutic trajectory following intra-articular hyaluronic acid injection in knee osteoarthritis – meta-analysis”. Osteoarthritis and Cartilage 19 (6): 611–619. doi:10.1016/j.joca.2010.09.014.PMID 21443958. edit
- Salk, R. S.; Chang, T. J.; d’Costa, W. F.; Soomekh, D. J.; Grogan, K. A. (2006). “Sodium Hyaluronate in the Treatment of Osteoarthritis of the Ankle: A Controlled, Randomized, Double-Blind Pilot Study”. The Journal of Bone and Joint Surgery 88 (2): 295–302.doi:10.2106/JBJS.E.00193. PMID 16452740. edit
- Beasley, K.; Weiss, M.; Weiss, R. (2009). “Hyaluronic Acid Fillers: A Comprehensive Review”.Facial Plastic Surgery 25 (2): 086–094. doi:10.1055/s-0029-1220647. PMID 19415575. edit
- Shimmura, S.; Ono, M.; Shinozaki, K.; Toda, I.; Takamura, E.; Mashima, Y.; Tsubota, K. (1995).“Sodium hyaluronate eyedrops in the treatment of dry eyes”. The British journal of ophthalmology 79 (11): 1007–1011. PMC 505317. PMID 8534643. edit
- Boucher, W. S.; Letourneau, R.; Huang, M.; Kempuraj, D.; Green, M.; Sant, G. R.; Theoharides, T. C. (2002). “Intravesical sodium hyaluronate inhibits the rat urinary mast cell mediator increase triggered by acute immobilization stress”. The Journal of Urology 167 (1): 380–384.doi:10.1016/S0022-5347(05)65472-9. PMID 11743360. edit

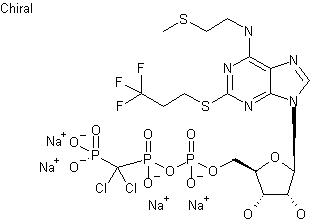
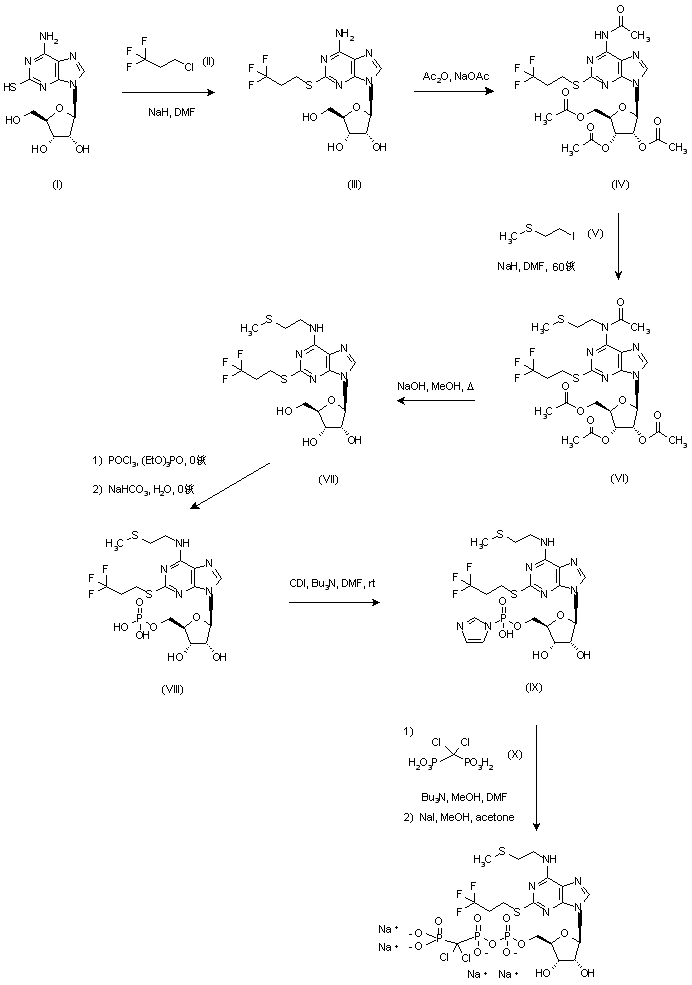
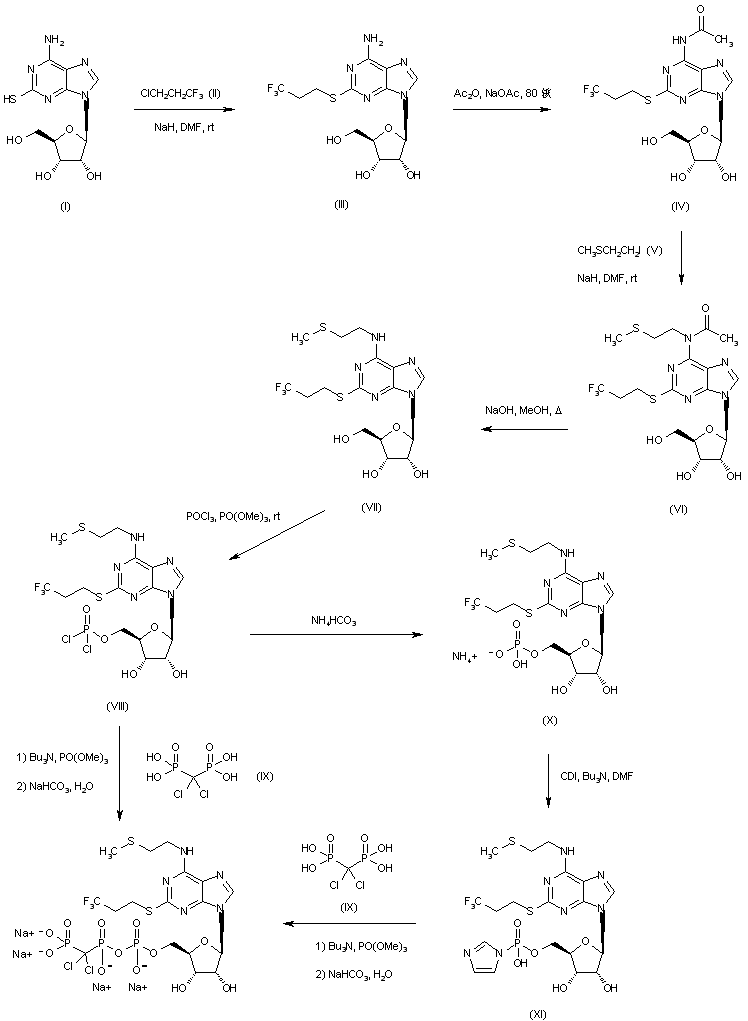
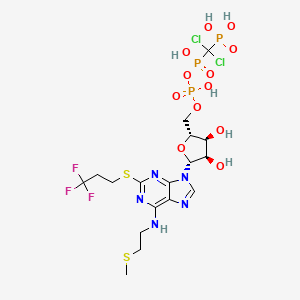 Cangrelor
Cangrelor


 OR
OR

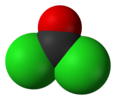
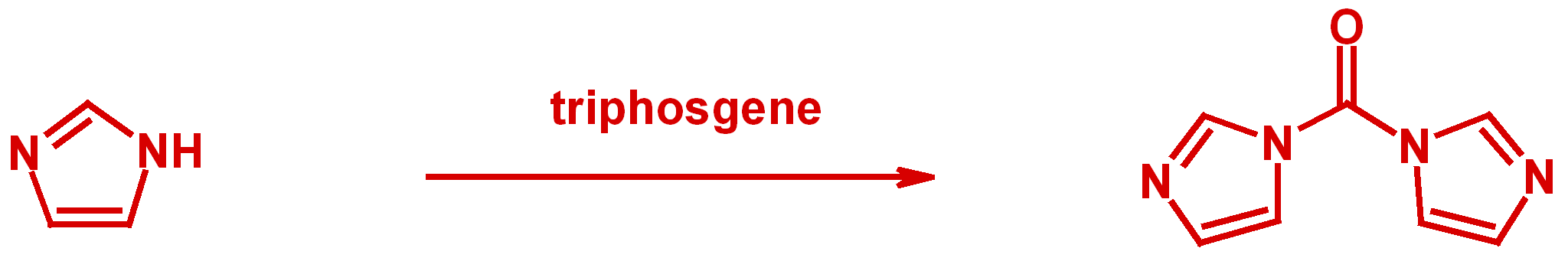
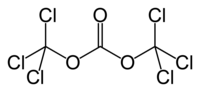 TRIPHOSGENE
TRIPHOSGENE








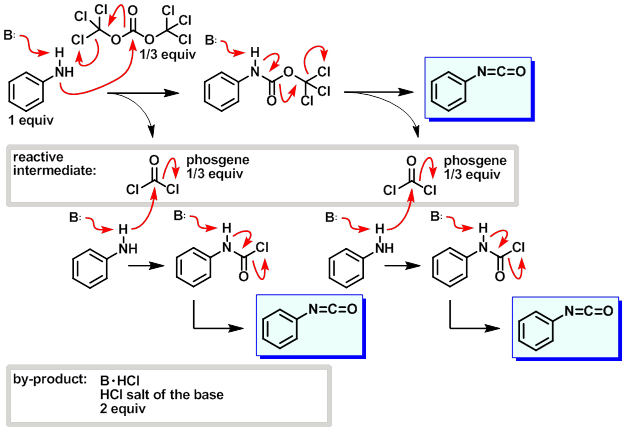
 lsu.edu
lsu.edu