Nabriva’s lefamulin receives FDA fast-track status to treat CABP and ABSSS
Austria-based Nabriva Therapeutics has received qualified infectious disease product (QIDP) and fast-track status designation from the US Food and Drug Administration for its lefamulin (BC 3781).
Austria-based Nabriva Therapeutics has received qualified infectious disease product (QIDP) and fast-track status designation from the US Food and Drug Administration for its lefamulin (BC 3781).
read
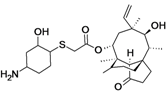
BC-3781
Topical pleuromutilin antibiotic agent
Gram-positive, including MRSA, PHASE 2 COMPLETED,Infection, acute bacterial skin and skin structure (ABSSSI)
Nabriva (Austria), Nabriva Therapeutics AG
BC-3781
cas 1061872-97-6
UNII-61H04Z5F9K
(3aS,4R,5S,6S,8R,9R,9aR,10R)-5-Hydroxy-4,6,9,10-tetramethyl-1-oxo-6-vinyldecahydro-3a,9-propanocyclopenta[8]annulen-8-yl [[(1R,2R,4R)-4-amino-2-hydroxycyclohexyl]sulfanyl]acetate;
14-O-[2-[(1R,2R,4R)-4-Amino-2-hydroxycyclohexylsulfanyl]acetyl]mutilin
BC-3781 is a pleuromutilin antibiotic in early clinical development at Nabriva for the treatment of community acquired pneumonia and for the treatment of patients with acute bacterial skin and skin structure infections (ABSSSI). Pleuromutilin antibiotics interfere with bacterial protein synthesis via a specific interaction with the 23S rRNA of the 50S bacterial ribosome subunit. They have a distinct antibacterial profile and show no cross-resistance with any other class of antibiotics. In 2012, a codevelopment agreement was signed between Forest and Nabriva, but, in 2014, this agreement terminated and Nabriva retained all rights.

The pleuromutilin BC-3781 belongs to the first generation of pleuromutilins to combine excellent oral
bioavailability with substantial activity against Gram-positive pathogens and atypicals as well as some
Gram-negative pathogens. In particular, BC-3781 is highly active against multi-drug resistant (MDR)
pathogens including methicillin resistant Staphylococcus aureus (MRSA), MDR Streptococcus pneumonia
(i.e. macrolide and quinolone resistance), and vancomycin resistant Enterococcus faecium. It is
characterized by excellent in vivo activities (e.g. pneumonia model), outstanding PK/PD parameters,
allowing once a day dosing, and a novel mode of action. BC-3781 is being developed for both oral and IV
administration and is intended for the treatment of serious multi-drug resistant skin & skin structure
infections (CSSI) and moderate to severe pneumonia (CAP, HAP etc).
Pleuromutilins have been known since 1951, but only entered the market in 2007 with the approval of retapamulin for topical use. Until today, there are no pleuromutilins for systemic use approved in human clinical practice.
in 2007 with the approval of retapamulin for topical use. Until today, there are no pleuromutilins for systemic use approved in human clinical practice.
Nabriva is currently working on the development of new compounds is this class. The lead compound, BC-3781, if approved, will be the first pleuromutilin for systemic use in humans.
The compound shows potent in vitro activity against a large collection of staphylococci, streptococci, andE. faecium. When compared to linezolid and vancomycin, the compound shows greater overall potency againstS. aureus [121]. BC-3781 shows improved activity against most bacteria commonly associated with community-acquired respiratory tract infections, the compound is especially potent against S. pneumoniaincluding penicillin resistant strains. It also shows improved activity against H. influenza, M. catarrhalis, M. pneumoniae and C. pneumoniae.
BC-3781 is undergoing Phase I clinical trials for CAP and in March of 2011 has completed a Phase II clinical study comparing it to vancomycin for treatment of aBSSSI [119,120,121,122,123]. Nabriva Therapeutics AG announced that the cooperation with Forest Laboratories to develop the compound had elapsed, and that Nabriva retained all rights in BC-3781. The company informed that the product was Phase III ready and that it was seeking partners to continue further development [203].
Nabriva is also developing BC-7013 for topical use against Gram-positive infections and working on the discovery of new pleuromutilins [119,124].
Dr William Prince, CMO Nabriva Therapeutics commented:
“This is the first patient study with a systemic pleuromutilin. It will be an important proof of concept
for an exciting new class of antibiotics. The phase II study builds on our extensive preclinical and
phase I data which have demonstrated that BC-3781 can achieve therapeutically relevant blood and
tissue levels in man with excellent tolerability when administered by either oral or intravenous
routes.”
“This is the first patient study with a systemic pleuromutilin. It will be an important proof of concept
for an exciting new class of antibiotics. The phase II study builds on our extensive preclinical and
phase I data which have demonstrated that BC-3781 can achieve therapeutically relevant blood and
tissue levels in man with excellent tolerability when administered by either oral or intravenous
routes.”
Dr. David Chiswell, CEO Nabriva Therapeutics commented:
“With a worldwide problem due to antibiotic resistant bacteria, there is a very significant need for
new classes of antibiotics with unique modes of action such as the pleuromutilins. The commercial
prospects for BC-3781 as the leading compound of an exciting new class are excellent, especially as it
has an ideal anti-bacterial spectrum for both skin and respiratory infections and is being developed
with both oral and intravenous formulations”
“With a worldwide problem due to antibiotic resistant bacteria, there is a very significant need for
new classes of antibiotics with unique modes of action such as the pleuromutilins. The commercial
prospects for BC-3781 as the leading compound of an exciting new class are excellent, especially as it
has an ideal anti-bacterial spectrum for both skin and respiratory infections and is being developed
with both oral and intravenous formulations”
BC-3781 is highly active against key pathogens, including MRSA, associated with skin infections and
community and hospital acquired pneumonia and is more potent than Linezolid and vancomycin. The
compound’s novel mode of action ensures that it overcomes resistance mechanisms affecting all
approved classes of antibiotics. BC-378
community and hospital acquired pneumonia and is more potent than Linezolid and vancomycin. The
compound’s novel mode of action ensures that it overcomes resistance mechanisms affecting all
approved classes of antibiotics. BC-378
About Nabriva Therapeutics
Nabriva Therapeutics is a biotechnology company focused on developing a new class of antibiotics for
the treatment of serious infections caused by resistant pathogens. Nabriva’s lead systemic product,
BC-3781, is being developed for the treatment of serious skin infections and bacterial pneumonia
caused by S. aureus, , S. pneumoniae, H. influenza, Mycoplasma, Legionella and other bacteria,
including drug resistant strains such as MRSA and vancomycin resistant E. faecium. In addition,
Nabriva Therapeutics’ topical pleuromutilin product candidate, BC-7013, is in clinical phase I. Nabriva
Therapeutics has a proven track record in world-class medicinal chemistry, clinical expertise, a
seasoned management team and solid IP. Nabriva Therapeutics is located in Vienna, Austria.
Nabriva Therapeutics is a biotechnology company focused on developing a new class of antibiotics for
the treatment of serious infections caused by resistant pathogens. Nabriva’s lead systemic product,
BC-3781, is being developed for the treatment of serious skin infections and bacterial pneumonia
caused by S. aureus, , S. pneumoniae, H. influenza, Mycoplasma, Legionella and other bacteria,
including drug resistant strains such as MRSA and vancomycin resistant E. faecium. In addition,
Nabriva Therapeutics’ topical pleuromutilin product candidate, BC-7013, is in clinical phase I. Nabriva
Therapeutics has a proven track record in world-class medicinal chemistry, clinical expertise, a
seasoned management team and solid IP. Nabriva Therapeutics is located in Vienna, Austria.
For more information on Nabriva please visit http://www.nabriva.com. Nabriva Therapeutics AG
…………………………………………
EP 2390245
……………………………………………..
The trivial name mutilin refers to the IUPAC systematic name (1S, 2R, 3S, 4S, 6R, 7R, 8R, 14R)-3,6-dihydroxy-2,4,7,14-tetramethyl-4-vinyl-tricyclo[5.4.3.01,8]tetradecan-9-one. In the examples, pleuromutilin derivatives are numbered in analogy to the mutilin numbering system described by H. Berner (Berner, H.; Schulz, G.; Schneider H. Tetrahedron 1980, 36, 1807-1811.):

…………………………………………………….
Pleuromutilin, a compound of formula A

is a naturally occurring antibiotic, e.g. produced by the basidomycetes Pleurotus mutilus and P. passeckerianus, see e.g. The Merck Index, 13th edition, item 7617. A number of further pleuromutilins having the principle ring structure of pleuromutilin and being substituted at the hydroxy group have been developed, e.g. as antimicrobials.
From WO 02/04414 Al pleuromutilin derivatives, e.g. 14-O-[(Aminocyclohexan-2-yl (and - 3-yl)-sulfanyl)-acetyl]-mutilins; from WO 07/014409 Al e.g. 14-O-[((Mono- or dialkylamino)-cycloalkylsulfanyl)-acetyl]-mutilins and from WO 07/000004 Al e.g. [((Acyl- hydroxy-amino)-cycloalkylsulfanyl)-acetyl]-mutilins, are known.
14-O-{[(1R, 2R, 4R)-4-Amino-2-hydroxy-cyclohexylsulfanyl]-acetyl}- mutilin hydrochloride
Example 1 – 14-O-{[(1R, 2R, 4R)-4-Amino-2-hydroxy-cyclohexylsulfanyl]-acetyl}- mutilin hydrochloride + (IS, 2S, 4S) diastereomer hydrochloride
Step Al. 14-O-{[(1R, 2R, 4R)-4-tert-Butoxycarbonylamino-2-hydroxy- cyclohexylsulfanyl]-acetyl}-mutilin + (IS, 2S, 4S) diastereomer and 14-O-{[(lR, 2R, 5S)-5-i'eri'-Butoxycarbonylamino-2-hydroxy-cyclohexylsulfanyl]-acetyl}- mutilin + (IS, 2S, 5R) diastereomer and
14-0-{[(lR, 2R, 4S)-4-tert-Butoxycarbonylamino-2-hydroxy-cyclohexylsuIfanyl]-acetyl}- mutilin + (IS, 2S, 4R) diastereomer
To a solution of 3,4-epoxycyclohexyl-carbamic acid tert-butyl ester (Gomez-Sanchez, E.; Marco-Contelles J. Tetrahedron 2005, 61, 1207-1219.) (4.27g, 20mmol) and pleuromutilin thiol (Nagarajan, R. Eli Lilly and Company 1978, US4, 130,709) (7.10 g, 18 mmol) in 200 ml of tetrahydrofuran was added aluminum oxide (40 g, Brockmann activity I, neutral) and the resulting mixture was stirred for 40 hours at room temperature. The suspension was filtered and concentrated under reduced pressure. The residue was subjected to chromatography (silica, cyclohcxane / ethyl acetate = 1/1) to yield 14-O-{[(1R, 2R, 4R)-4-ler(- butoxycarbonylamino-2-hydroxy-cyclohcxylsulfanyl]-acctyl}-mutilin + (IS, 2S, 4S) diastereomer (a) (Rf = 0.38, 1.34g, 12%) as well as a mixture of 14-O-{[(1R, 2R, 5S)-5-tert- butoxycarbonylumino-2-hy(lroxy-cyclohcxylsulfnnyl]-ncctyl}-niυtilin + (I S, 2S, 5R) diastereomer and 14-O-{[(1R, 2R, 4S)-4-tert-butoxycarbonylamino-2-hydroxy- cyclυhexylsulfanyl]-acetyl}-mutilin + (IS, 2S, 4R) diastereomer (b) (Rf = 0.26, 2.81 g, 25%) as colorless amorphous foams. (a): 1H NMR (400MHz, DMSOd6, δ, ppm, inter alia): 6.74 (d, IH, NH, J = 7Hz), 6.13 (dd, IH, 19-H, J – I lHz and 18Hz), 5.54 (d, IH, 14-H, J = 8Hz), 5.05 (m, 2H, 20-H), 4.90 (d, IH, 2′-OH, J = 5Hz), 4.48 (d, IH, 11-OH, J = 6Hz), 3.55 – 3.20 (m, 6H, 1 ‘-H, 2‘-H, 4′-H, 11-H, 22-H), 2.40 (bs, IH, 4-H), 1.36 (s, 3H, 15-CH3), 1.35 (s, 9H, tert-butyl), 1.06 (s, 3H, 18-CH3), 0.81 (d, 3H, 17-CH3, J = 7Hz), 0.62 (d, 3H, 16-CH3, J = 7Hz). MS-ESI (m/z): 630 (MNa+), 1237 (2MNa+).
(b): 1H NMR (400MHz, DMSO-de, δ, ppm, inter alia): 6.70 (d, IH, NH, J = 7Hz), 6.12 (dd, IH, 19-H, J = HHz and 18Hz), 5.34 (d, IH, 14-H, J = 8Hz), 5.05 (m, 2H, 20-H), 4.82, 4.78 (d, IH, 2′-OH, J = 4Hz), 4.48 (d, IH, 11-OH, J = 6Hz), 3.55 – 3.20 (m, 5H, 2′-H, 475′-H, 11- H, 22-H), 2.97 (m, IH, 1 ‘-H), 2.40 (bs, IH, 4-H), 1.35 (s, 12H, 15-CH3, tert-butyl), 1.05 (s, 3H, 18-CH3), 0.82 (d, 3H, 17-CH3, J = 7Hz), 0.62 (d, 3H, 16-CH3, J = 7Hz). MS-ESI (m/z): 630 (MNa+), 1237 (2MNa+).
or Step A2. 14-O-{[(1R, 2R, 4R)-4-tert-Butoxycarbonylamino-2-liydroxy- cyclohexylsulfanyl]-acetyl}-mutilin + (IS, 2S, 4S) diastereomer and
14-O-{[(1R, 2R, 5S)-5-tert-Butoxycarbonylamino-2-hydroxy-cyclohexylsulfanyl]-acetyl}- mutilin + (IS, 2S, 5R) diastereomer and
14-O-{[(1R, 2R, 4S)-4-rerf-Butoxycarbonylamino-2-hydroxy-cyclohexylsulfanyl]-acetyl}- mutilin + (IS, 2S, 4R) diastereomer
To a solution of 3,4-epoxycyclohexyl-carbamic acid tert-butyl ester (10 g, 47 mmol) and pleuromutilin thiol (16.6 g, 42 mmol) in 200 ml of methanol and 20 ml of dioxane was added 2N NaOH (21 ml, 42 mmol) and the resulting mixture was stirred for 16 hours at room temperature. After completion of the reaction the pH was set to 7 with diluted HCl and the reaction mixture was concentrated under reduced pressure. The residue was diluted with water and brine and extracted three times with ethyl acetate. The organic layers were dried over sodium sulfate and filtered. The filtrate was concentrated under reduced pressure and after chromatography (silica, cyclohexane / ethyl acetate = 1/1) 14-O-{[(1R, 2R, 4R) A-tert- butoxycarbonylamino-2-hydroxy-cyclohexylsulfanyl] -acetyl }-mutilin + (IS, 2S, 4S) diastereomer (Rf = 0.40, 3.1g, 12% yield) as well as a mixture of 14-O-{[(1R, 2R, 5S)-5-tert- butoxycarbonylamino-2-hydroxy-cyclohexylsulfanyl]-acetyl}-mutilin + (IS, 2S, 5R) diastereomer and 14-O-{[(1R, 2R, 4S)-4-tert-butoxycarbonylamino-2-hydroxy- cyclohexylsulfanyl]-acetyl}-mutilin + (IS, 2S, 4R) diastereomer (Rf = 0.25, 6.35 g, 25%) were obtained as colorless amorphous foams. or Step A3. 14-O-{[(1R, 2R, 4R)-4-tert-Butoxycarbonylamino-2-hydroxy- cyclohexylsulfanyl]-acetyl}-mutilin + (IS, 2S, 4S) diastereomer and 14-O-{ [(1R, 2R, 5S)-5-tert-Butoxycarbonylamino-2-hydroxy-cyclohexylsulfanyl]-acetyl}- mutilin + (IS, 2S, 5R) diastereomer
To a solution of Pleuromutilin thiol (9.25 g, 23.5 mmol) in 100 ml of acetonitrile (dried over 4A molecular sieve) was added l,5-diazabicyclo[4.3.0]non-5-ene (DBN, 2.9 μl, 23.5 mmol) and after 1 hour of stirring at room temperature under argon atmosphere the mixture was ^ charged with syn-3,4-epoxycyclohexyl-carbamic acid tert-butyl ester (4.17 g, 19.5 mmol) and stirred for further 16 hours at room temperature. The reaction mixture was concentrated under reduced pressure. The residue was charged with water and brine and extracted three times with dichloromethane. The organic layers were dried over sodium sulphate and filtered. The filtrate was concentrated under reduced pressure and subjected to chromatography (silica, cyclohexane / ethyl acetate = 1/1) to yield 14-O-{[(1R, 2R, 4R)-4-teAY-butoxycarbonylamino- 2-hydroxy-cyclohexylsulfanyl]-acetyl}-mutilin + (IS, 2S, 4S) diastereomer (Rf = 0.38, 5.07g, 43%) as well as 14-O-{[(1R, 2R, 5S)-5-tert-butoxycarbonylamino-2-hydroxy- cyclohexylsulfanyl]-acetyl}-mutilin + (IS, 2S, 5R) diastereomer (Rf = 0.25, 2.95 g, 16.5%) as colorless amorphous foams.
Step B. 14-O-{[(1R, 2R, 4R)-4-Amino-2-hydroxy-cyclohexylsulfanyl]-acetyl}-mutilin + (IS, 2S, 4S) diastereomer
To a solution of 14-O-{[(1R, 2R, 4R)-4-teΛ-t-butoxycarbonylamino-2-hydroxy- cyclohexylsulfanyl]-acetyl}-mutilin + (IS, 2S, 4S) diastereomer (1.34 g, 2.20 mmol) in 75 ml of dichloromethane was added trifiuoroacetic acid (4 ml) at 4°C and stirred for 5 hours at room temperature. The reaction mixture was diluted with dichloromethane and cautiously poured into a saturated NaHCO3 solution. The phases were separated and the aqueous layer was washed two times with dichloromethane. The combined organic layers are dried over sodium sulfate and filtered. After chromatography (silica, ethyl acetate/methanol/35% ammonia solution = 50/50/1) 14-O-{[(1R, 2R, 4R)-4-amino-2-hydroxy-cyclohexylsulfanyl]- acetyl}-mutilin + (IS, 2S, 4S) diastereomer (745 mg, 67% yield) was obtained as colorless amorphous foam.
1H NMR (400MHz, DMSO-de, δ, ppm, inter alia): 6.14 (dd, IH, 19-H, J = 1 IHz and 18Hz), 5.54 (d, IH, 14-H, J = 8Hz), 5.05 (m, 2H, 20-H), 4.50 (d, IH, 11-OH, J = 6Hz), 3.50 – 3.20 (m, 5H, 2′-H, 4′-H, H-H, 22-H), 2.55 (m, IH, l ‘-H), 2.40 (bs, IH, 4-H), 1.35 (s, 3H, 15- CH3), 1.06 (s, 3H, 18-CH3), 0.82 (d, 3H, 17-CH3, J = 7Hz), 0.62 (d, 3H, 16-CH3, J = 7Hz). MS-ESI (m/z): 508 (MH+), 530 (MNa+), 1015 (2MH+), 1037 (2MNa+).
Step C. 14-O-{[(1R, 2R, 4R)-4-Amino-2-hydroxy-cyclohexylsulfanyI]-acetyl}- mutilin hydrochloride + (IS, 2S, 4S) diastereomer hydrochloride
A solution of 14-O-{[(1R, 2R, 4R)-4-amino-2-hydroxy-cyclphexylsulfanyl] -acetyl }-mutilin + (IS, 2S, 4S) diastereomer (325 mg, 0.64 mmol) in 20 ml of dioxane was treated with IN HCl (0.64ml, 0.64 mmol). After stirring at room temperature for 30 minutes the solution was lyophilized to obtain 14-O-{[(1R, 2R, 4R)-4-amino-2-hydroxy-cyclohexylsulfanyl] -acetyl }- mutilin hydrochloride + (IS, 2S, 4S) diastereomer hydrochloride (quantitative yield) as colorless amorphous solid.
1H NMR (500MHz, DMSO-Cl6, δ, ppm, inter alia): 7.6 (bs, 3H, NH3 +), 6.14 (dd, IH, 19-H, J = 1 IHz and 18Hz), 5.55 (d, IH, 14-H, J = 8Hz), 5.05 (m, 2H, 20-H), 4.52 (d, IH, H-OH, J = 6Hz), 3.50 – 3.20 (m, 4H, 2′-H, H-H, 22-H), 3.03 (m, IH, 4′-H), 2.53 (m, IH, 1 ‘-H), 2.40 (bs, IH, 4-H), 1.37 (s, 3H, 15-CH3), 1.06 (s, 3H, 18-CH3), 0.82 (d, 3H, 17-CH3, J = 7Hz), 0.62 (d, 3H, 16-CH3, J = 7Hz). MS-ESI (m/z): 508 (MH+), 530 (MNa+), 1015 (2MH+), 1037 (2MNa+), 542 (MCl“).
Example IA – 14-O-{[(1S, 2S, 4S)-4-Amino-2-hydroxy-cyclohexylsulfanyl]-acetyl}- mutilin and 14-O-{[(1R, 2R, 4R)-4-Amino-2-hydroxy-cyclohexylsuIfanyl]-acetyl}-mutilin
The mixture of 14-O-{[(1R, 2R, 4R)-4-Amino-2-hydroxy-cyclohexylsulfanyl]-acetyl}-mutilin + (IS, 2S, 4S) diastereomer (12 g, 23.6 mmol) from Example 1 Step B was separated on a cbiral column (250 x 20 mm CHIRALCEL OD-H, n-heptane / ethanol / diethylamine = 80/20/0.1) to yield 14-O-{[(1S*, 2S*, 4S*)-4-amino-2-hydroxy-cyclohexylsulfanyl]-acetyl}- mutilin (a) (early eluting compound, 4.76 g, 37% yield, uncorrected) and 14-O-{[(1R*, 2R*, 4R*)-4-amino-2-hydroxy-cyclohexylsulfanyl]-acetyl}-mutilin (b) (late eluting compound, 3.63 g, 30% yield, uncorrected) as colorless amorphous foams.
(a): 1H NMR (400MHz, DMSO-(I6, δ, ppm, inter alia): 6.13 (dd, IH, 19-H, J = 1 IHz and 18Hz), 5.54 (d, IH, 14-H, J = 8Hz), 5.05 (m, 2H, 20-H), 4.50 (d, IH, H-OH, J = 6Hz), 3.50 – 3.20 (m, 5H, 2′-H, 4′-H, H-H, 22-H), 2.55 (m, IH, l ‘-H), 2.40 (bs, IH, 4-H), 1.35 (s, 3H, 15- CH3), 1.05 (s, 3H, 18-CH3), 0.82 (d, 3H, 17-CH3, J = 7Hz), 0.62 (d, 3H, 16-CH3, J = 7Hz). MS-ESI (m/z): 508 (MH+), 530 (MNa+), 1015 (2MH+), 1037 (2MNa+), 506 (M-H) “, 542 (MCl“).
(b): 1H NMR (400MHz, DMSO-d6> δ, ppm, inter alia): 6.13 (dd, IH, 19-H, J = 1 IHz and 18Hz), 5.54 (d, IH, 14-H, J = 8Hz), 5.05 (m, 2H, 20-H), 4.50 (d, IH, H-OH, J = 6Hz), 3.50 – 3.20 (m, 5H, 2′-H, 4′-H, 11-H, 22-H), 2.55 (m, IH, 1 ‘-H), 2.40 (bs, IH, 4-H), 1.35 (s, 3H, 15- CH3), 1.05 (s, 3H, 18-CH3), 0.82 (d, 3H, 17-CH3, J = 7Hz), 0.62 (d, 3H, 16-CH3, J = 7Hz). MS-ESI (m/z): 508 (MH+), 530 (MNa+), 1015 (2MH+), 1037 (2MNa+), 506 (M-H) “, 542 (MCl“).
…………………………………………………….
WO 2011146954
The present invention relates to crystalline 14-0-{[(4-amino-2-hydroxy-cyclohexyl)- sulfanyl] -acetyl }-mutilin, new processes for its preparation and crystalline salts thereof.
Pleuromutilin, a compound of formula
Pleuromutilin

is a naturally occurring antibiotic, e.g. produced by the basidiomycetes Pleurotus mutilus and P. passeckerianus, see e.g. The Merck Index, 12th edition, item 7694.
A number of further pleuromutilins having the principle ring structure of pleuromutilin and being substituted at the primary hydroxy group have been developed, e.g. as antimicrobials. Due to their pronounced antimicrobial activity, a group of pleuromutilin derivatives, amino- hydroxy-substituted cyclohexylsulfanylacetylmutilins, as disclosed in WO 2008/113089, have been found to be of particular interest. As described in WO2008/11089 14-0-{[(4- Amino-2-hydroxy-cyclohexyl)-sulfanyl] -acetyl }-mutilins are particularly useful compounds because they demonstrate activity against Gram-positive and Gram-negative pathogens e.g. associated with respiratory tract and skin and skin structure infections. For the production of substantially pure isomers/diastereomers of this group of compounds, there is a need for a production process which is convenient for use on an industrial scale and which also avoids the use of costly starting materials, environmentally hazardous reagents and solvents or time consuming and laborious purification steps. The production process described in WO 2008/113089 involves chromatographic purification of the compounds prepared according to individual synthesis steps and the final diastereomers are separated by chiral HPLC chromatography which cannot be used on industrial scale. Surprisingly, crystalline intermediates have been found which on the one hand have unexpected chemical purification potential which is important for the production processes for pure amino-hydroxy-substituted cyclohexylsulfanylacetylmutilins avoiding
chromatographic purification and separation steps.
It has to be pointed out that 14-0-{[(4-amino-2-hydroxy-cyclohexyl)-sulfanyl]-acetyl}- mutilins are potential new drug substances for the human market with regulatory
requirements defined in the corresponding ICH guidelines (International Conference on Harmonization of Technical Requirements for Registration of Pharmaceuticals for Human Use). The ICH guideline on impurities in new drug substances (Q3 A(R2)) includes the following thresholds:

As can be seen from the ICH thresholds above it is desirable to have all individual unknown impurities below 0.10% area and the structure elucidated impurities below 0.15%, respectively. Processes provided according to the present invention enable to produce APIs (Active Pharmaceutical Ingredients) within the desired specifications and fulfilling ICH requirements.
On the other hand, even more surprisingly, the crystalline intermediates yields to significant chiral enrichment which has a huge benefit in the production of the pure stereoisomers starting from cheaper racemic materials or less chirally pure starting materials. The described processes do not involve any chromatographic purification neither normal nor chiral phase in contrast to the synthetic procedures described in WO2008/113089 wherein is disclosed e.g. in Example 1, Step B that 14-0-{[(4-amino-2-hydroxy-cyclohexyl)-sulfanyl]- acetyl}-mutilins was isolated in the form of diastereomeric mixtures as colorless amorphous foams after normal phase chromatography. The chiral pure diastereomers are described to have been received in WO2008/113089, e.g. in Example 1 A after subjecting the mixture to chiral chromatography whereafter the separated pure diastereomers were isolated in the form of colorless amorphous foams.
Chiral chromatography, however is not a technology which can be applied on industrial large scale, and moreover no crystalline salts of 14-0-{[(4-amino-2-hydroxy-cyclohexyl)- sulfanyl]-acetyl}-mutilins were obtained according to WO2008/113089. In contrast to that, according to the present invention crystalline pharmaceutical acceptable salts of 14-0-{[(4-amino-2-hydroxy-cyclohexyl)-sulfanyl]-acetyl}-mutilins having surprising and superior properties over the amorphic prior art salts disclosed in
WO2008/113089 have been found; e.g. surprisingly the chemical stability of the crystalline salts of the present invention is improved over the amorphic salt forms; and also and in addition the crystalline salts of the present invention show a surprising low hygroscopicity.
Processes for the preparation of such crystalline salts wherein the salts may be obtained in a single stereoisomeric form from 14-0-{[(4-amino-2-hydroxy-cyclohexyl)-sulfanyl]-acetyl}- mutilins and processes for the preparation of stereoisomerically pure 14-0-{[(4-amino-2- hydroxy-cyclohexyl)-sulfanyl]-acetyl}-mutilins in crystalline form as a basis for the crystalline salts have also been found.
In one aspect the present invention provides a process for the preparation of a compound of formula I

in the form of a single stereoisomer in crystalline form, comprising
deprotecting the amine group
either in a compound of formula Ila

in a mixture of a compound of formula Ila with a compound of formula lib

wherein R is an amine protecting group, and isolating a compound of formula I obtained in the form of a single diastereomer in crystalline form either directly from the reaction mixture or via recrystallization in organic solvent.
In another aspect the present invention provides a compound of formula I as defined above in the form of a single stereoisomer in crystalline form.
Compounds of formula Ila are new and also form part of the present invention.
In another aspect the present invention provides a compound of formula Ha.
In a compound of formula I, or Ha, respectively, the carbon atoms of the cyclohexyl ring to which the hydroxy group, the amine group and the sulfanyl-acetyl-mutilin group are attached are all in the R configuration and thus a compound of formula I, or Ila represents an optionally amine protected 14-0-{[(l ?,2i?,4 ?)-4-amino-2-hydroxy-cyclohexylsulfanyl]- acetyl}-mutilin. In contrast to that, in a compound of formula lb

or lib the carbon atoms of the cyclohexyl ring to which the hydroxy group, the amine group and the sulfanyl-acetyl-mutilin group are attached are all in the S configuration and thus a compound of formula lib represents an optionally amino protected 14-0-{[(lS,2S,4S)-4-Amino-2-hydroxy- cyclohexylsulfanyl] -acetyl } -mutilin. An amine protecting group includes protecting groups known to a skilled person and which are removable under acidic, basic, hydrogenating, oxidative or reductive methods, e.g. by hydrogenolysis, treatment with an acid, a base, a hydride, a sulfide. Appropriate amine protecting groups e.g. are described in T. W. Greene, P. G. M. Wuts, Protective Groups in Organic Synthesis, Wiley-Interscience, 4th edition, 2007, particularly p. 696-868.
Example 1
tert-Butyl [(lR3Ri4R)-3-hydroxy-4-mercapto-cyclohexyl]-carbamate

3.94 Kg of {(li?,2J?,4i?)-4-[(tert-Butoxycarbonyl)-amino]-2-hydroxy-cyclohexyl}-benzene- carbothioate and 37 L of CH2C12 were charged to a vessel and the mixture obtained was stirred at 15-25°C. 0.39 Kg of 1,4-dithio-DL-threitol (10% wt) was added to the mixture and rinsed through with 2 L of CH2C12. To the mixture obtained 0.84 Kg of hydrazine monohydrate was added. The mixture obtained was stirred at 18 to 22°C for 3 h and the reaction was followed by HPLC. Upon completion of the reaction, 39 L of 1 M phosphoric acid solution was added and the mixture obtained was stirred for a further 15-30 min. Two phases formed were separated and the organic phase obtained was washed with 39 L of of 1 M phosphoric acid solution followed by 39 L 1% aqueous NaCl solution. The organic layer obtained was concentrated in vacuo at <40°C, to the concentration residue 20 L of CH2C12 was added and the mixture obtained again was concentrated. To the concentration residue obtained a further 8 L of CH2C12 was added and the mixture obtained was concentrated to dryness.
2.89 Kg of tert-Butyl [(l ?,3/?,4i?)-3-hydroxy-4-mercapto-cyclohexyl]-carbamate in the form of a white solid was obtained.
1H NMR (200 MHz, DMSO-de, ppm) δ 6.79 (d, J=7.8Hz, 1H), 4.99 (d, J=5.8Hz, 1H), 3.34 – 3.24 (m, 1H), 3.14 – 3.04 (m, 1H), 2.37 (d, J=3.8 Hz, 1H), 2.00 -1.89 (m, 1H), 1.87 – 1.82 (m, 1H), 1.73 – 1.67 (m, 1H), 1.47 – 1.04 (m, 12H)
Example 2
22-0-TosylpIeuromutilin

22-O-Tosylpleuromutilin is a known compound from literature. However a preparation procedure is outlined below.
A solution of 13.0 kg of pleuromutilin and 6.57 kg of 4-toluenesulfonyl chloride in 42.1 L of CH2CI2 at 10 to 15 °C was treated with 9.1 L of 5.7 M aqueous NaOH over 20 min, maintaining a temperature < 25 °C. The resulting off-white suspension was heated to reflux for 20 h and the reaction was followed until completion determined by HPLC. Upon reaction completion the mixture obtained was cooled to 20 to 30 °C, diluted with 52 L of CH2C12, stirred at 15 to 25 °C for 10 min, and the layers obtained were separated. The organic phase obtained was washed several times with 52 L of water until a pH of the aqueous layer was adjusted to < 9. The organic layer obtained was concentrated to 4 volumes and
azeotropically dried twice with 52 L of CH2C12. To the solution obtained 52 L of heptane were added dropwise and the solution obtained was concentrated at < 40 °C to
approximately 4 volumes. To the concentrate obtained 52 L of heptane was added and the resulting suspension was stirred at 20 to 25 °C for 2 to 2.5 h, filtered, the filter cake obtained was washed with 39 L of heptane and pulled dry on the filter.
The solid was dried under vacuum at < 40 °C for at least 12 h.
16.9 kg of 22-O-tosylpleuromutilin in the form of a white solid was obtained.
1H NMR (200 MHz, DMSO-d6, ppm, inter alia) δ 7.81 (d, 2H), 7.47 (d, 2H), 6.14 – 6.0 (m, 1H), 5.54 (d, J=7.8Hz, 1H), 5.08 – 4.99 (m, 2H), 4.70 (AB, J=16.2Hz, 2H), 3.41 (d, J=5.2Hz, 1H), 2.41(s, 4H), 1.04(s, 3H), 0.81 (d, 3H), 0.51 (d, 3H)
Example 3
14-0-{[(l/?,2R,4R)-4-ter/-Butoxycarbonylamino-2-hydroxy-cyclohexyI-sulfanyl]-acetyl}- mutilin

4.75 Kg of Pleuromutilin tosylate (Tos-PLEU) and 44.4 L of MTBE were charged into a vessel and to the mixture obtained 0.31 Kg of benzyl-tri-«-butylammonium chloride was added and rinsed through with 2.4 L of MTBE. To the mixture obtained 20 L of IM aqueous NaOH solution and 2.84 Kg of tert-Butyl [(lif,3i?,4^)-3-hydroxy-4-mercapto-cyclohexyl]- carbamate were added and the mixture obtained was stirred at 17 to 23 °C for 3 h. Upon completion of the reaction (determined by HPLC) two layers formed were separated and the lower aqueous layer was removed. The organic phase obtained was washed with 19 L of IM aqueous NaOH solution, twice with 20 L of 0.1 M phosphoric acid, 20 L of 10% aqueous NaHC03 solution and twice with 20 L of water. The organic liquors obtained were concentrated, the concentrate obtained was taken up in 7.46 Kg of 2-propanol, the mixture obtained was concentrated again and dried in vacuo at <40°C. 6.66 Kg of 14-O- { [( 1 -¾,2i?,4i?)-4-/ert-Butoxycarbonylamino-2-hydroxy-cyclohexyl-sulfanyl]-acetyl } -mutilin in the form of a white foam was obtained.
Ή NMR (200 MHz, DMSO-d6, ppm, inter alia) δ 6.78 (d, J=7.8Hz, 1H), 6.22 – 6.08 (m,lH), 5.55 (d, J=7.8Hz, 1H), 5.13 – 5.02 (m, 2H), 4.95 (d, J=5Hz, 1H), 4.52 (d, J=6Hz, 1H), 3.36 (AB, J=15Hz, 2H), 2.40 (s, broad, 1H), 2.15 – 2.0 (m, 3H), 1.9 – 1.8 (m, 1H), 1.35 (s, 9H), 0.81 (d, J=7Hz, 3H), 0.62 (d, J=6.6Hz, 3H)
MS (ESI, g/mol): m/z 653 [M+2Na] +
Example 4
14-0-{[(lR,2R,4R)-4-Amino-2-hydroxy-cyclohexyIsulfanyI]-acetyl}-mutilin, crystalline Form 2

Step A: 14-O-{[(li?.2i?,4i?)-4-Amino-2Thvdroxy-cyclohexylsulfanvn-acetvU-mutilin in crystalline Form 1
6.6 Kg of 14-O-{[(li?,2if,4/?)-4-tert-Butoxycarbonylamino-2-hydroxy-cyclohexyl-sulfanyl]- acetyl}-mutilin and 13.2 L of isopropanol were charged into a vessel and stirred at 20 to 25°C. 11.20 kg of 85% phosphoric acid was added and the mixture obtained was heated to approximately 50°C for at least 16 h. The mixture obtained was analyzed for reaction completion by HPLC. Upon completion of the reaction the mixture was cooled to 20 to 25°C and 52 L of CH2C12 was added. The mixture obtained was cooled to 0 to 5°C and 51 L of 30% aqueous K2CO3 solution was added over 1 h at <25°C. The mixture obtained was warmed to rt, stirred for 30 min and the pH of the aqueous layer was determined. To the mixture obtained a further 15 L of 30% aqueous K2C03solution was added at <25°C, the mixture obtained was stirred at 15°C to 25 °C for 30 min and the two phases obtained were separated. The aqueous phase obtained was extracted with 51 L of CH2CI2 and the combined organic phases were washed with 51 L of purified water. The mixture obtained was concentrated to a volume of 25 L, 33.6 Kg of CH2C12 was added and the mixture obtained was concentrated to 25 L. To the concentrate obtained 33.6 Kg of CH2C12 was added and the mixture obtained was concentrated to 10 L. The concentration residue obtained was cooled to 18 to 22°C and 50 L of di-wopropyl ether was added over a period of 1 h. The slurry obtained was stirred at 15 to 25°C for a minimum of 2 h, filtered and the solid obtained was washed with 10 L of di-wopropyl ether and was dried.
3.79 Kg of 14-0-{[(li?,2i?,4if)-4-Amino-2-hydroxy-cyclohexylsulfanyl]-acetyl}-mutilin in crystalline Form 1 was obtained.
Step B: 14-O-{r(l-R.2i?,4j?)-4-amino-2-hvdroxy-cvclohexylsulfanyl1-acetv -mutilin. in crystalline Form 2
For further purification 14-O-{[(l ?,2 ?,4i?)-4-Amino-2-hydroxy-cyclohexylsulfanyl]- acetyl}-mutilin from Step A and 18.75 L of n-butanol were heated to 88 to 92°C until complete dissolution and stirred for 30 to 60 min. The mixture obtained was allowed to cool to 40 to 45°C over at least 2 h and further stirred at this temperature for 2 h. The mixture obtained was filtered and the precipitate obtained was washed with 3.75 L of «-butanol followed by 3.75 L of MTBE. That purification procedure was repeated and the resultant product was dried in vacuo at <40°C.
3.27 Kg of crystalline 14-0-{[(li?,2if,4i?)-4-amino-2-hydroxy-cyclohexylsulfanyl]-acetyl}- mutilin in crystalline Form 2 was obtained in the form of a white solid.
lH NMR (400 MHz, CDC13, ppm, inter alia) δ 6.51 – 6.44 (m, 1H), 5.78 (d, J=8Hz, 1H), 5.38 – 5.20 (m, 2H), 3.48 – 3.40 (m, 1H), 3.36 (d, J=7Hz, 1H), 3.25 (AB, J=15Hz, 2H), 2.92 – 2.82 (m, 1H), 2.6 – 2.5 (m, 1H), 1.45 (s, 3H), 1.20 (s, 3H), 0.88 (d, J=7Hz, 3 H), 0.73 (d, J=8Hz, 3H)
MS (ESI, g/mol): m/z 508 [M+H] +
Example 5
14-0-{[(lR^/f,4R)-4-Amino-2-hydroxy-cyclohexylsulfanyl]-acetyl}-mutilin, crystalline

To a solution of 900 g of 14-0-{[(li?,2i?,4i?)-4-tert-butoxycarbonylamino-2-hydroxy- cyclohexyl-sulfanyl]-acetyl}-mutilin in 9 L of CH2C12 at 15 to 25°C was added 1.8 L of TFA at 15 to 25°C and the resulting solution was stirred for 2 h. Following reaction completion the reaction mixture was concentrated under vacuum and the concentration residue obtained was azeo-dried with a total of 9 L of CH2C12. The concentrate obtained was dissolved in 4.5 L of CH2C12, the solution obtained cooled to 0 to 5°C and the pH was adjusted to pH 11 with aqueous 3.6 L 2CO3 (2.5M) solution. The biphasic mixture obtained was warmed to 15 to 20°C and stirred for 5 to 10 minutes. The layers obtained were separated, the aqueous phase obtained was extracted with 1.8 L of CH2C12, the organic phases obtained were combined, washed with 2.3 L of H20, dried over Na2S04 and concentrated to dryness under vacuum at <40°C. Crude 14-0- { [( 1 R,2R,4R)-4- Amino-2-hydroxy-cyclohexyl-sulfanyl]-acetyl } -mutilin was obtained. Yield: 744 g
For further purification the following procedure was applied:
To 744 g of crude 14-O-{[(li?,2i?,4i-)-amino-2-hydroxy-cyclohexyl-sulfanyl]-acetyl}- mutilin was added 2.23 L of THF and the resulting suspension was stirred at 15 to 25°C for 60 min. To the mixture obtained 7.44 L of MTBE was added over 15 to 30 min, the suspension obtained was aged for 60 min and filtered under nitrogen. The collected solids were washed with a total of 3 L of MTBE and pulled dry on the filter under nitrogen for 1.5 h.
626 g of 14-0-{[(li?,2i?,4i?)-4-Amino-2-hydroxy-cyclohexyl-sulfanyl]-acetyl}-mutilin in crystalline Form 1 was obtained.
The Ή NMR pattern confirms the structure of 14-O-{[(li?,2i?,4i?)-4-amino-2-hydroxy- cyclohexylsulfanyl] -acetyl} -mutilin. The NMR pattern for 14-O-{[(l ?,2i?,4/?)-4-amino-2- hydroxy-cyclohexylsulfanyl]-acetyl}-mutilin is described in example 4.
…………………………………………………………
REF
119
Nabriva. Pleuromutilins. Available online: http://www.nabriva.com/programs/pleuromutilins/ (accessed on 7 December 2012).
120
120
Forest Laboratories. Our pipeline: Solid, and set for further growth. Available online: http://www.frx.com/research/pipeline.aspx (accessed on 13 April 2013).
121
121
Sader, H.S.; Biedenbach, D.J.; Paukner, S.; Ivezic-Schoenfeld, Z.; Jones, R.N. Antimicrobial activity of the investigational pleuromutilin compound BC-3781 tested against Gram-positive organisms commonly associated with acute bacterial skin and skin structure infections. Antimicrob. Agents Chemother. 2012,56, 1619–1623, doi:10.1128/AAC.05789-11.
122
Sader, H.S.; Paukner, S.; Ivezic-Schoenfeld, Z.; Biedenbach, D.J.; Schmitz, F.J.; Jones, R.N. Antimicrobial activity of the novel pleuromutilin antibiotic BC-3781 against organisms responsible for community-acquired respiratory tract infections (CARTIs). J. Antimicrob. Chemother. 2012, 67, 1170–1175, doi:10.1093/jac/dks001.
Sader, H.S.; Paukner, S.; Ivezic-Schoenfeld, Z.; Biedenbach, D.J.; Schmitz, F.J.; Jones, R.N. Antimicrobial activity of the novel pleuromutilin antibiotic BC-3781 against organisms responsible for community-acquired respiratory tract infections (CARTIs). J. Antimicrob. Chemother. 2012, 67, 1170–1175, doi:10.1093/jac/dks001.
123
Nabriva Therapeutics AG. Study comparing the safety and efficacy of two doses of BC-3781 vs. vancomycin in patients with acute bacterial skin and skin structure infection (ABSSSI). Available online: http://www.clinicaltrials.gov/ct2/show/NCT01119105 (accessed on 13 April 2013).
Nabriva Therapeutics AG. Study comparing the safety and efficacy of two doses of BC-3781 vs. vancomycin in patients with acute bacterial skin and skin structure infection (ABSSSI). Available online: http://www.clinicaltrials.gov/ct2/show/NCT01119105 (accessed on 13 April 2013).
124
Novak, R. Are pleuromutilin antibiotics finally fit for human use? Ann. NY Acad. Sci. 2011, 1241, 71–81, doi:10.1111/j.1749-6632.2011.06219.x.
Novak, R. Are pleuromutilin antibiotics finally fit for human use? Ann. NY Acad. Sci. 2011, 1241, 71–81, doi:10.1111/j.1749-6632.2011.06219.x.
 valnemulin
valnemulin retapamulin
retapamulin| WO2002004414A1* | Jul 9, 2001 | Jan 17, 2002 | Gerd Ascher | Pleuromutilin derivatives having antibacterial activity |
| WO2007000004A1* | Jun 26, 2006 | Jan 4, 2007 | Nabriva Therapeutics Forschung | Pleuromutilin derivatives containing a hydroxyamino- or acyloxyaminocycloalkyl group |
| WO2007014409A1* | Jul 26, 2006 | Feb 8, 2007 | Nabriva Therapeutics Forschung | Pleuromutilin derivatives useful as antibacterials |
| WO2008011089A2 | Jul 19, 2007 | Jan 24, 2008 | Mark B Gagner | Wagering game with special-event eligibility feature based on passive game play |
| WO2008113089A1 | Mar 19, 2008 | Sep 25, 2008 | Nabriva Therapeutics Ag | Pleuromutilin derivatives for the treatment of diseases mediated by microbes |
| US20060276503 * | Aug 30, 2004 | Dec 7, 2006 | Glaxo Group Limited | Novel process salts compositions and use |
No comments:
Post a Comment