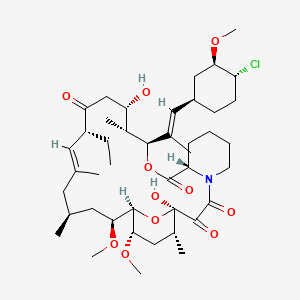
(3S,4R,5S,8R,9E,12S,14S,15R,16S,18R,19R,26aS)- 3-{(E)-2-[(1R,3R,4S)-4-Chloro-3-methoxycyclohexyl]- 1-methylvinyl}-8-ethyl-5,6,8,11,12,13,14,15,16,17,
18,19,24,25,26,26a-hexadecahydro-5,19-dihydroxy- 14,16-dimethoxy-4,10,12, 18-tetramethyl-15,19-epoxy- 3H-pyrido[2,1-c][1,4]oxaazacyclotricosine-1, 7,20,21(4H,23H)-tetrone
The systematic name of pimecrolimus is (lR,9S,12S,13R,14S,17R,18E,21S,23S,24R,25S,27R)-12-[(lE)-2- {(1 R,3R,4S)-4-chloro-3-methoxycyclohexyl} - 1 -methylvinyl] - 17-ethyl- 1,14- dihydroxy-23,25-dimethoxy-13,19,21,27-tetramethyl-ll,28-dioxa-4-aza- tricyclo[22.3.1.04'9]octacos-18-ene-2,3,10,16-tetraone.
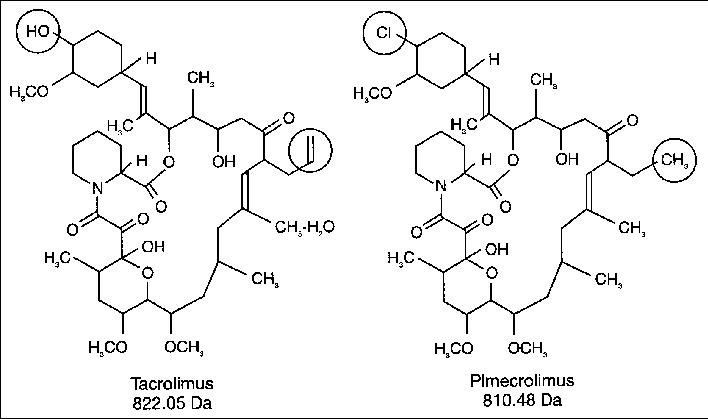
Pimecrolimus is the 32 epichloro derivative of ascomycin.
Elidel, NCGC00167506-01, DSSTox_CID_26674, DSSTox_RID_81811, DSSTox_GSID_46674, 137071-32-0, Tox21_112504
Pimecrolimus is an immunomodulating agent used in the treatment of atopic dermatitis (eczema). It is currently available as a topical cream, once marketed by Novartis, (however Galderma will be promoting the molecule in Canada in early 2007) under the trade name Elidel.
Pimecrolimus is a chemical that is used to treat atopic dermatitis (eczema). Atopic dermatitis is a skin condition characterized by redness, itching, scaling and inflammation of the skin. The cause of atopic dermatitis is not known; however, scientists believe that it may be due to activation of the immune system by various environmental or emotional triggers. Scientists do not know exactly how pimecrolimus reduces the manifestations of atopic dermatitis, but pimecrolimus reduces the action of T-cells and mast cells which are part of the immune system and contribute to responses of the immune system. Pimecrolimus prevents the activation of T-cells by blocking the effects of chemicals (cytokines) released by the body that stimulate T-cells. Pimecrolimus also reduces the ability of mast cells to release chemicals that promote inflammation.
Ascomycin macrolactams belong to a new group of immunosuppressive, immunomodulatory and anti-inflammatory agents and include, e.g., ascomycin (FK520), tacrolimus (FK506) and pimecrolimus (ASM 981). The main biological effect of ascomycin macrolactams appears to be the inhibition of the synthesis of both Th1 and Th2-type cytokines in target cells.
As used herein, the term “ascomycin macrolactam” means ascomycin, a derivative of ascomycin, such as, e.g., tacrolimus and pimecrolimus, or a prodrug or metabolite of ascomycin or a derivative thereof.
Ascomycin, also called immunomycin, is a structurally complex macrolide produced by Streptomyces hygroscopicus. Ascomycin acts by binding to immunophilins, especially macrophilin-12. It appears that ascomycin inhibits the production of Th1 (interferon- and IL-2) and Th2 (IL-4 and IL-10) cytokines. Additionally, ascomycin preferentially inhibits the activation of mast cells, an important cellular component of the atopic response. Ascomycin produces a more selective immunomodulatory effect in that it inhibits the elicitation phase of allergic contact dermatitis but does not impair the primary immune response when administered systemically. The chemical structure of ascomycin is depicted below.
Tacrolimus (FK506) is a synthetic derivatives of ascomycin. As a calcineurin inhibitor, it works through the FK-binding protein and inhibits the dephosphorylation of nuclear factor of activated T cells (NFAT), thereby preventing the transport of the cytoplasmic component of NFAT to the cell nucleus. This leads to transcriptional inhibition of proinflammatory cytokine genes such as, e.g., interleukin 2, which are dependent on the nuclear factor of activated NFAT. The chemical structure of tacrolimus is depicted below.
Pimecrolimus, an ascomycin derivative, is a calcineurin inhibitor that binds with high affinity to the cytosolic receptor macrophilin-12, inhibiting the calcium-dependent phosphatase calcineurin, an enzyme required for the dephosphorylation of the cytosolic form of the nuclear factor of the activated T cell (NF-AT). It thus targets T cell activation and proliferation by blocking the release of both TH1 and TH2 cytokines such as IF-g, IL-2, -4, -5, and -10.3 It also prevents the production of TNF-a and the release of proinflammatory mediators such as histamine, hexosaminidase, and tryptase from activated mast cells.3 It does not have general antiproliferative activity on keratinocytes, endothelial cells, and fibroblasts, and in contrast to corticosteroids, it does not affect the differentiation, maturation, functions, and viability of human dendritic cells. The chemical structure of pimecrolimus is depicted below.
Pimecrolimus is an anti-inflammatory compound derived from the macrolactam natural product ascomycin, produced by certain strains of Streptomyces.
Pimecrolimus binds with high affinity to macrophilin-12 (FKBP-12) and inhibits the calcium-dependent phosphatase, calcineurin. As a consequence, it inhibits T cell activation by blocking the transcription of early cytokines. In particular, pimecrolimus inhibits at nanomolar concentrations Interleukin-2 and interferon gamma (Th1-type) and Interleukin-4 and Interleukin-10 (Th2-type) cytokine synthesis in human T cells. Also, pimecrolimus prevents the release of inflammatory cytokines and mediators from mast cells in vitro after stimulation by antigen/lgE.
ELIDEL® (pimecrolimus) Cream 1% contains the compound pimecrolimus, the immunosuppressant 33-epi-chloro-derivative of the macrolactam ascomycin.
Chemically, pimecrolimus is (1R,9S,12S,13R,14S,17R,18E,21S,23S,24R,25S,27R)-12-[(1E)-2{(1R,3R,4S)-4-chloro-3-methoxycyclohexyl}-1-methylvinyl]-17-ethyl-1,14-dihydroxy-23,25 dimethoxy-13,19,21,27-tetramethyl-11,28-dioxa-4-aza-tricyclo[22.3.1.0 4,9]octacos-18-ene2,3,10,16-tetraone.
The compound has the empirical formula C43H68CINO11 and the molecular weight of 810.47. The structural formula is
Pimecrolimus is a white to off-white fine crystalline powder. It is soluble in methanol and ethanol and insoluble in water.
Each gram of ELIDEL Cream 1% contains 10 mg of pimecrolimus in a whitish cream base of benzyl alcohol, cetyl alcohol, citric acid, mono- and di-glycerides, oleyl alcohol, propylene glycol, sodium cetostearyl sulphate, sodium hydroxide, stearyl alcohol, triglycerides, and water.
The second representative of the immunosuppressive macrolides for topical application - after tacrolimus (Protopic ®) - has 21 October in the trade. Pimecrolimus is approved for short-term and intermittent long-term treatment for patients aged two years who suffer from mild to moderate atopic dermatitis.
Pimecrolimus is a lipophilic derivative of macrolactam Ascomycin. The macrolides inhibit the production and release of pro-inflammatory cytokines by blocking the phosphatase calcineurin.The anti-inflammatory effect unfolds the drug in the skin. Since he is only minimally absorbed to not measurable, it hardly affects the local or systemic immune response. Therefore, the authorization neither restricts nor a maximum daily dose treatable area or duration of therapy.The cream can also be applied on the face, head and neck, and in skin folds, but not simultaneously with other anti-inflammatory topical agents such as glucocorticoids.
In studies in phases II and III patients aged three months and treated a maximum of one year.In two six-week trials involving 186 infants and young children as well as 403 children and adolescents, the verum symptoms and itching decreased significantly better than the cream base. Already in the first week of itching in 44 percent of children and 70 percent of the infants improved significantly. In adults, pimecrolimus was less effective than 0.1 percent betamethasone 17-valerate.
In the long-term treatment the verum significantly reduced the incidence of flares, revealed two studies with 713 and 251 patients. About a half and one year each about twice as many of the small patients were free of acute disease exacerbations than with the cream base (example: 61 versus 34 per cent of children, 70 versus 33 percent of infants older than six months). Moreover, the use of topical corticosteroids decreased significantly.
In a study of 192 adults with moderate to severe eczema half suffered six months no relapses more (24 percent with placebo). In the long-term therapy pimecrolimus was less effective than 0.1 percent triamcinolone acetonide cream and 1 percent hydrocortisone cream in adults.
The new topicum is-apart from burning and irritation at the application site - relatively well tolerated. It is neither kontaktsensibilisierend still phototoxic or sensitizing and does not cause skin atrophy. As in atopic Ekzen but usually a long-term therapy is necessary studies can reveal long-term adverse effects of the immunosuppressant on the skin only beyond one year.Also available from direct comparative studies between tacrolimus and pimecrolimus. They could help to delineate the importance of the two immunosuppressants.
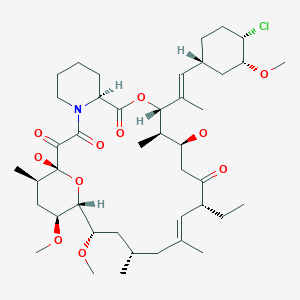
Pimecrolimus (registry number 137071-32-0; Figure 1) is a macro lide having anti-inflammatory, antiproliferative and immunosuppressive properties. This substance is present as an active ingredient in the Elidel ® drug recently approved in Europe and in the USA for topical treatment of inflammatory conditions of the skin such as atopic dermatitis.
Figure 1: structural formula of pimecrolimus
19th Ed., vol. π, pg. 1627, spray-drying consists of bringing together a highly dispersed liquid and a sufficient volume of hot air to produce evaporation and drying of the liquid droplets. Spray-drying however is often limited to aqueous solutions unless special expensive safety measures are taken. Also, in spite of the short contact time, certain undesirable physical and chemical characteristics of the emerging solids are in particular cases unavoidable. The turbulence present in a spray-drier as a result of the moving air may alter the product in an undesirable manner. Modifications to the spray-drying technique are disclosed in WO 03/063821 and WO 03/063822. [00012] European Patent EP 427 680 Bl discloses a method of synthesizing amorphous pimecrolimus (Example 66a). The method yields amorphous pimecrolimus as a colorless foamy resin.
U.S. Patent No. US 6,423,722 discloses crystalline forms of pimecrolimus, such as form A, form B, etc. US 722 also contend that by performing example 66a from the European Patent EP 427 680 Bl, amorphous pimecrolimus is obtained.
The preparation of pimecrolimus was described for the first time in the patent application EP427680 on behalf of Sandoz. Used as raw material in such document is ascomycin (compound identified by registry number 11011-38-4), a natural product obtained through fermentation from Streptomyces strains (such as for example Streptomyces hygroscopicus var ascomyceticus, or Streptomyces hygroscopicus tsukubaensis N°9993). Pimecrolimus is obtained from the ascomycin through a sequence of four steps of synthesis (scheme 1)
Scheme 1 : synthesis process described in EP427680
From a structural point of view, pimecrolimus is the 33-epi-chloro derivative of ascomycin. As described in EP427680, the simultaneous presence - in the structure of ascomycin - of two secondary hydroxyl groups in position 24 and in position 33, requires the protection of the hydroxyl in position 24 before substituting the second hydroxyl in position 33 with an atom of chlorine.
In order to obtain the monoprotection of the hydroxyl in position 24 of ascomycin, such synthesis process provides for the preparation of 24,33-disilyl derivative and the subsequent selective removal of the silyl ester in position 33.
The high ratio between the silylating agent and the substrate and the non-complete selectivity of the subsequent step of deprotection requires carrying out two chromatographic purifications on the column of silica gel (Baumann K., Bacher M., Damont A., Hogenauer K., Steck A. Tetrahedron, (2003), 59, 1075-1087). The general yields of such synthesis process are not indicated in literature; an experiment by the applicant revealed that such yields amount to about 16% molar starting from ascomycin.
Other synthesis processes were recently proposed as alternatives to the synthesis of EP427680.
In particular, the International patent application WO2006040111 on behalf of Novartis provides for the direct substitution of the hydroxyl in position 33 of ascomycin with an atom of chlorine and a second alternative, described in the international patent application WO2006060614 on behalf of Teva, uses - as a synthetic intermediate - a sulfonate derivative in position 33 of ascomycin. Both the proposed synthetic alternatives are not entirely satisfactory in that in WO2006040111 the proposed halogenating agents (chlorophosphorane and N- chlorosuccinimide) are not capable, according to the same authors, of regioselectively substituting the hydroxyl function in position 33, while in WO2006060614 the quality characteristics of the obtained product are, even after chromatographic purification and/or crystallisation, low for a product to be used for pharmaceutical purposes (i.e. purity of 96% as described in the experimental part).
Generally, purified enzymatic systems may be used for the organic synthesis of polyfunctional molecules (Wang Y-F, Wong C-H. J Org Chem (1988) 53, 3127- 3129; Santaniello E., Ferraboschi P., Grisenti P., Manzocchi A. Chem. Rev. (1992), 92(5), 1071-140; Ferraboschi P., Casati S., De Grandi S., Grisenti P., Santaniello E. Biocatalysis (1994), 10(1-4), 279-88); WO2006024582). WO2007103348 and WO2005105811 describe the acylation of rapamycin in position 42 in the presence of lipase from Candida antartica.
.........................
Scheme 2: synthesis of pimecrolimus for enzymatic transesterification of ascomycin.
Scheme 3. Synthesis of pimecrolimus for enzyme-catalyzed alcoholysis from 33,24- diacetate of ascomycin
Example 1
Preparation of the 33-acetyl derivative of ascomvcin (compound I of scheme II)
Lipase from Candida antarctica (CAL B, Novozym 435) [0.140 g (2 U/mg)
FLUKA] was added to a solution of ascomycin (100 mg; 0.126 mmol) in toluene (8 ml) and vinyl acetate (4.5 eq; 0.473 g). The reaction is kept under stirring at the temperature of 30° C for 80 hrs then the enzyme is taken away for filtration and the filtrate is concentrated at low pressure to obtain 105 mg of 33-acetyl ascomycin.
A sample of such intermediate was purified for analytical purposes by chromatography on silica gel (n-hexane/acetone = 8/2 v/v as eluents) and thus crystallised by acetone/water.
The following analysis were carried out on such sample: 1H-NMR (500MHz) δ:
2.10 (CH3CO), 3.92 and 4.70 (24CH and 33CH); IR (cm-1): 3484.245, 2935.287,
1735.331, 1649.741, 1450.039,
1372.278; DSC: endotherm at 134.25° C; [α]D=-74,0° (c=0.5 CHCl3).
Spectrum of MS (ESI +): m/z: 856.4 (M+23; 100.0%)
Elementary analysis calculated for C45H7iNO13: C 64.80%; H, 8.58%; N, 1.68%;
O, 24.94%
Elementary analysis found: C 64.78%; H, 8.54%; N, 1.59%; O, 24.89%
Preparation of the 24-tgrt-butyldimethylsilylether-33 -acetyl derivative of ascomvcin (intermediate 24-silyl-33-Oac; compound II of scheme 2)
2,6-lutidine (0.29Og; 2.7 mmolels) and tert-butyldimethylsilyl triflate (0.238g; 0.9 mmoles) are added to a solution of 33-acetyl derivative of ascomycin (150 mg;
0.18 mmoles) in dichloromethane (5ml). The reaction is left under stirring at ambient temperature for 30 minutes. After this period the reaction mixture is washed with a solution saturated with sodium bicarbonate (5 ml) and organic phase obtained is washed in sequence with HCl 0.1N (5 ml 3 times) and with a solution at 30% of NaCl (5ml). The organic phase is anhydrified on sodium sulphate, filtered and concentrated to residue under vacuum to obtain 128 mg of product.
Spectrum of MS (ESI +): m/z: 970.5 (M+23; 100.0%)
1H-NMR (500 MHz) δ: 0.05 and 0.06 ((CHs)2Si), 0.90 ((CH3)3C-Si), 2.10
(CH3CO), 4.70 (33CH)
IR (cm-'): 3462.948, 2934.450, 1739.236, 1649.937
Elementary analysis calculated for C51H85NOi3Si: C 64.59%; H, 9.03%; N, 1.48%; O, 21.93%
Elementary analysis found: C 64.50%; H, 9.05%; N, 1.41%; O, 21.88%
DSC= endoderma a 236,43° C. [α]D=-81,4° (c=0.5 CHCl3).
Preparation of 24-tert-butyldimethylsilylether of ascomycin (intermediate 24- silyl-33-OH; compound III of scheme 2) n-octan-1-ol (0.035g; 0.265 mmoles) and CAL B (Novozym 435) [0.100 g (2
U/mg) FLUKA] are added to a solution of 24-tert-butyldimethylsilylether-33- acetyl derivative of ascomycin (50 mg; 0.053 mmoles) in tert-butylmethylether (4 ml). The reaction is kept under stirring at the temperature of 40° C for 120 hours.
After this period the reaction mixture is filtered and the filtrate is evaporated to residue under vacuum to obtain a reaction raw product which is purified by chromatography on silica gel: 44 mg of product (0.048 mmoles) are recovered through elution with petroleum ether/acetone 7/3.
The chemical/physical properties of the obtained product match those of a reference sample obtained according to patent EP427680.
Preparation of 24-tert-butyldimethylsilylether-33-epi-chloro ascomycin
(intermediate 24-silyl-33-chloro; compound IV of scheme 2)
A solution of 24-silyl FR520, i.e. 24-silyl ascomycin (165 g; 0.18 moles) in anhydrous toluene (1.4 litres) and pyridine (50 ml) is added to a suspension of dichlorotriphenylphosphorane (99.95g) in anhydrous toluene (1.1 litres), under stirring at ambient temperature (20-25 °C) in inert atmosphere.
After adding, the reaction mixture is heated at the temperature of 60° C for 1 hour.
After this period the temperature of the reaction mixture is taken to 25° C and thus the organic phase is washed in sequence with water (1 time with 1 L) and with an aqueous solution of NaCl at 10% (4 times with 1 L each time), then it is anhydrified on sodium sulphate, filtered and concentrated under vacuum to obtain about 250 g of a moist solid of toluene. Such residue product is retaken with n- hexane (500 ml) and then evaporated to dryness (in order to remove the toluene present). The residue product is diluted in n-hexane (500 ml) under stirring at ambient temperature for about 45 minutes and then the undissolved solid taken away for filtration on buckner (it is the sub-product of dichlorophosphorane).
The filtrate is concentrated at low pressure to obtain 148.6 g of a solid which is subsequently purified by chromatography on silica gel (elution with n- heptane/acetone = 9/1) to obtain 123 g (0.13 moles) of product.
The chemical/physical properties of the obtained product match those described in literature (EP427680).
Preparation of the pimecrolimus from 24-fert-butyldimethylsilylether-33-epi- chloro ascomycin
The intermediate 24-silyl-33 chloro (123g; 0.13 Moles; compound IV of scheme
2) is dissolved under stirring at ambient temperature in a dichloromethane/methanol mixture=l/l=v/v (1.1 litres) then p-toluenesulfonic acid monohydrate (10.11 g) is added.
The reaction is kept under stirring at the temperature of 20-25° C for 72 hours, thus a solution of water (600 ml) and sodium bicarbonate (4.46 g) is added to the reaction mixture. The reaction mixture is kept under stirring at ambient temperature for 10 minutes, the organic phase is then prepared and washed with an aqueous solution at 10% of sodium chloride (600 ml).
The organic phase is anhydrified on sodium sulphate, filtered and concentrated under vacuum to obtain 119 g of raw pimecrolimus. Such raw product is purified by chromatography on silica gel (n-hexane/acetone as eluents) and thus crystallised by ethyl acetate, cyclohexane/water to obtain 66 g (81.5 mmoles) of purified pimecrolimus.
The chemical/physical data obtained matches the data indicated in literature.
Example 2
Preparation of ascomvcin 24.33-diacetate (intermediate 24, 33-diacetate; compound V of scheme 3)
DMAP (4.5 eq; 0.136 g) and acetic anhydride (4.5 eq; 0.114 g) are added to a solution of ascomycin (200 mg; 0.25 mmoles) in pyridine (2.5 ml), under stirring at the temperature of 0° C.
The reaction is kept under stirring for 1.5 hours at the temperature of 0° C then it is diluted with water and it is extracted with ethyl acetate (3 times with 5 ml). The organic extracts are washed with HCl 0.5 N (5 times with 10 ml), anhydrified on
Na2SO4 concentrated under vacuum.
The residue product was purified by chromatography on silica gel (n- hexane/acetone 8/2 v/v as eluent) to obtain ascomycin 24,32-diacetate (210 mg;
0.24 mmoles).
We carried out the following analysis on such purified sample:
1H-NMR (500 MHz) δ: 2.02 and 2.06 (2 CH3CO), 5.20 and 4.70 (24CH and
33CH);
IR (Cm-1): 3462.749, 2935.824, 1734.403, 1650.739, 1449.091, 1371.079.
DSC: endothermic peak at 234.10° C ; [α]D=- 100.0° (C=0.5 CHCl3).
Spectrum of MS (ESI+): m/z: 898.4 (100.0%; m+23).
Elementary analysis calculated for C47H73NO14: C 64.44%; H 8.40%; N 1.60%; O
25.57%
Elementary analysis found: C 64.55%; H 8.44%; N 1.61%; O 25.40%
Preparation of the 24-acetyl ascomycin (intermediate 24-acetate-33-OH; compound VI of scheme 3)
Lipase from Candida antartica (CAL B Novozym 435) [1.1 g (2 U/mg) FLUKA] is added to a solution of ascomycin 33,24-diacetate (500 mg; 0.57 mmol) in
TBDME (25 ml) and n-octan-1-ol (4.5 eq; 0.371 g). The reaction is kept under stirring at 30° C for 100 hours, then the enzyme is taken away for filtration and the obtained filtrate is concentrated under low pressure to obtain 425 mg (0.51 mmoles) of product.
A sample was purified for analytical purposes by chromatography on silica gel (n- hexane/acetone = 7:3 v/v as eluents) and thus crystallised by acetone/water.
We carried out the following analysis on such purified sample: 1H-NMR
(500MHz) δ: 2.05 (CH3CO); IR (an 1): 3491.528, 2935.860, 1744.728, 1710.227,
1652.310, 1448.662, 1371.335. DSC: endothermic peak at 134.68° C; [α]D=-
102.7° (c=0.5 CHCl3)
Spectrum of MS (ESI +): m/z: 856.4 (M+23; 100.0%)
Elementary analysis calculated for C45H71NO13: C 64.80%; H, 8.58%; N, 1.68%;
0, 24.94%
Elementary analysis found: C 64.71%; H, 8.49%; N, 1.60%; O, 24.97%
Preparation of the 24-acetyl-33epi-chloro ascomycin (intermediate 24-Acetate-33- chloro; compound VII of scheme 3) Supported triphenylphosphine (0.335 g; 1.1 mmoles) is added to a solution of 24- acetyl ascomycin (400 mg; 0.48 mmoles) in carbon tetrachloride (5 ml). The reaction mixture is kept under reflux for 3 hours then it is cooled at ambient temperature. The obtained suspension is filtered and the filtrate is concentrated to residue under vacuum to obtain 0.45g of reaction raw product which is purified by chromatography on silica gel: 163mg (0.19 mmoles) of product are obtained by elution with petroleum ether/acetone = 90/10.
1H-NMR δ: 2.08 (CH3CO); 4.60 (33CH); IR (Cm"1)= 3464.941, 2934.360,
1738.993, 1650.366, 1450.424, 1371.557; DSC: endothermic peak at 231.67° C
[α]D=-75.2° (c=0.5 CHCl3)
Spectrum of MS (ESI +): m/z: 874.3 (M+23; 100.0%)
Elementary analysis calculated for C45H70ClNO12: C 63.40%; H, 8.28%; Cl,
4.16%; N, 1.64%; O, 22.52%
Elementary analysis found: C 63.31%; H, 8.30%; Cl, 4.05%; N, 1.58%; O,
22.42%.
Preparation of pimecrolimus from 24-acetyl-33-epi-chloro ascomycin
A solution of 24-acetyl-33-epi-chloro ascomycin (200 mg; 0.23 mmoles; compound VII) in methanol (2 ml) and HCl 3N (1 ml) is stirred at ambient temperature for 40 hours. After this period, the reaction is neutralised with an aqueous bicarbonate solution, the methanol evaporated under vacuum. The mixture is extracted with dichloromethane (3 times with 5 ml), anhydrified on sodium sulphate, filtered and concentrated to residue to obtain a residue product which is purified by chromatography on silica gel (n-hexane/acetone as eluents) and thus crystallised by ethyl acetate, cyclohexane/water to obtain 78 mg of purified pimecrolimus (0.096 mmoles).
The chemical/physical characteristics of the obtained product matches the data indicated in literature for pimecrolimus.
Example 4 (comparative*)
Verification of the method of synthesis of pimecrolimus described in EP427680 Imidazole (508 mg) and tert-Butyldimethylsilylchloride (1.125 g) are added in portions to a solution of 2g (2.53 mmoles) of ascomycin in anhydrous N,N- dimethylformamide (40 ml). The reaction mixture is kept under stirring at ambient temperature for 4.5 days. The reaction is thus processed diluting it with ethyl acetate (200 ml) and processing it using water (5 x 100 ml). The organic phase is separated, anhydrified on sodium sulphate, filtered and evaporated to residue under vacuum to obtain a foamy raw product which is subsequently purified by chromatography on silica gel (1:30 p/p): 2.1 g (2.05 mmoles; yields 81% molars) of ascomycin 24,33 disilyl intermediate are obtained by elution with n- hexane/ethyl acetate 3/1. The chemical/physical data of such intermediate matches that indicated in EP427680.
2.1 g (2.05 mmoles) of ascomycin 24,33 disilyl intermediate are dissolved in a solution under stirring at the temperature of 0°C composed of acetonitrile (42 ml) and aqueous HF 40% (23.1 ml). The reaction mixture is kept under stirring at the temperature of 0°C for 2 hours then it is diluted with dichloromethane (30 ml). Then the reaction is washed in sequence with a saturated aqueous solution using sodium bicarbonate (30 ml) and water (30 ml). The separated organic phase is anhydrified on sodium sulphate, filtered and evaporated to residue under vacuum to obtain a foamy residue which is subsequently purified by chromatography on silica gel (1:30 p/p): 839 mg (0.92 mmoles; yields 45% molars) of ascomycin 24 monosilyl intermediate are obtained by elution with dichloromethane/methanol 9/1. The chemical/physical data of such intermediate matches that obtained on the compound III scheme 2 and matches the data of literature indicated in EP427680. A mixture of 839 mg (0.92 mmoles; yields 45% molars) of ascomycin 24 monosilyl intermediate, triphenylphosphine (337 mg) in carbon tetrachloride (36.4 ml) is heated under stirring under reflux for 15 hours. After this period the reaction mixture is evaporated to residue under vacuum to obtain a solid product purified by chromatography on silica gel (1:30 p/p): 535 mg (0.57 mmoles; yields 63% molars) of ascomycin 24 monosilyl intermediate, 33-chloro derivative are obtained by elution with n-hexane/ethyl acetate 2/1. The chemical/physical data of such intermediate matches those we obtained on compound IV scheme 2 and matches the data of literature indicated in EP427680.
535 mg (0.57 mmoles) of ascomycin 24 monosilyl intermediate, 33-chloro derivative are dissolved under stirring at ambient temperature in acetonitrile (16.4 ml) and aqueous HF 40% (0.44 ml). The reaction mixture is kept under stirring at ambient temperature for 45' and then it is diluted with ethyl acetate (100 ml). The organic phase is thus washed in sequence with an aqueous solution of sodium bicarbonate (70 ml) with water (2 x 70 ml) and thus it is anhydrified on sodium sulphate, filtered and evaporated under vacuum to obtain a solid which is subsequently purified by chromatography on silica gel (1 :30 p/p): 323 mg (0.399 mmoles; yields 70% molars) of pimecrolimus is obtained by elution with n- hexane/ethyl acetate 2/3. The chemical/physical characteristics of the obtained product matches the data indicated in literature regarding pimecrolimus; the overall yield of the process is 16%.
.............................
Example 7: Preparation of amorphous pimecrolimus by precipitation [00094] 19,5 g purified pimecrolimus (colorless resin) was dissolved in 217 ml acetone at 4O0C and concentrated. Residue: 38,76 g. The residue was diluted with 6 ml distilled water with stirring. Finally 1 ml acetone was added. This solution was added slowly to 2 L chilled distilled water that was stirred efficiently. After the addition had been completed, the suspension was stirred 20 min at O0C. Then the solid was filtered and dried at 450C in vacuum oven overnight. Product: 15,65 g yellowish solid. Amorphous (XRD, DSC).
[00095] Procedure of grinding: 200 mg of Pimecrolimus sample was ground gently in an agate mortar using a pestle for half a minute. ,