
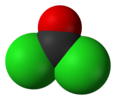
Phosgene
Phosgene is the chemical compound with the formula COCl2. This colorless gas gained infamy as a chemical weapon during World War I. It is also a valued industrial reagent and building block in synthesis of pharmaceuticals and other organic compounds. In low concentrations, its odor resembles freshly cut hay or grass.[3] In addition to its industrial production, small amounts occur naturally from the breakdown and the combustion oforganochlorine compounds, such as those used in refrigeration systems.[4] The chemical was named by combining the Greek words 'phos' (meaning light) and genesis (birth); it does not mean it contains any phosphorus (cf. phosphine).
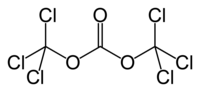 TRIPHOSGENE
TRIPHOSGENE
Triphosgene (bis(trichloromethyl) carbonate (BTC), C3Cl6O3) is a chemical compound that is used as a safer substitute for phosgene, because at room temperature it is a solid crystal, as opposed to phosgene which is a gas.Triphosgene crystals decompose above 200 °C READ .......http://www.buss-ct.com/e/company/publications/reaction_technology/eckert_reprint_CO6_2011-hr2.pdf
This compound is commercially available. It is prepared by exhaustive free radical chlorination of dimethyl carbonate:
- CH3OCO2CH3 + 3 Cl2 → CCl3OCO2CCl3 + 6 HCl
Triphosgene can be easily recrystallized from boiling hexanes to yield pure white crystals.
Triphosgene is used as a reagent in organic synthesis for a variety of chemical transformations including to bond one carbonyl group to two alcohols, and to convert an amine group into isocyanate.
The toxicity of triphosgene is the same as phosgene since it decomposes to phosgene on heating and upon reaction with nucleophiles. Even trace moisture leads to formation of phosgene. Therefore this reagent can be safely handled if one takes all the precautions as for phosgene.
Structure and basic properties
Phosgene is a planar molecule as predicted by VSEPR theory. The C=O distance is 1.18 Å, the C—Cl distance is 1.74 Å and the Cl—C—Cl angle is 111.8°.[5] It is one of the simplest acid chlorides, being formally derived from carbonic acid.
Industrially, phosgene is produced by passing purified carbon monoxide and chlorine gas through a bed of porous activated carbon, which serves as acatalyst:[4]
- CO + Cl2 → COCl2 (ΔHrxn = −107.6kJ/mol)
The reaction is exothermic, therefore the reactor must be cooled. Typically, the reaction is conducted between 50 and 150 °C. Above 200 °C, phosgene reverts to carbon monoxide and chlorine, Keq (300K) = 0.05. World production of this compound was estimated to be 2.74 million tonnes in 1989.[4]
Because of safety issues, phosgene is often produced and consumed within the same plant, and extraordinary measures are made to contain this toxic gas. It is listed on schedule 3 of the Chemical Weapons Convention: All production sites manufacturing more than 30 tonnes per year must be declared to the OPCW.[6] Although less dangerous than many other chemical weapons, such as sarin, phosgene is still regarded as a viablechemical warfare agent because it is so easy to manufacture when compared to the production requirements of more technically advanced chemical weapons such as the first-generation nerve agent tabun.[7]

Upon ultraviolet (UV) radiation in the presence of oxygen, chloroform slowly converts into phosgene by a radical reaction. To suppress thisphotodegradation, chloroform is often stored in brown-tinted glass containers. Chlorinated compounds used to remove oil from metals, such as automotive brake cleaners, are converted to phosgene by the UV rays of arc welding processes.[8]
Phosgene may also be produced during testing for leaks of older-style refrigerant gases. Chloromethanes (R12, R22 and others) were formerly leak-tested in situ by employing a small gas torch (propane, butane or propylene gas) with a sniffer tube and a copper reaction plate in the flame nozzle of the torch. If any refrigerant gas was leaking from a pipe or joint, the gas would be sucked into the flame via the sniffer tube and would cause a colour change of the gas flame to a bright greenish blue. In the process, phosgene gas would be created due to the thermal reaction. No valid statistics are available, but anecdotal reports suggest that numerous refrigeration technicians suffered the effects of phosgene poisoning due to their ignorance of the toxicity of phosgene, produced during such leak testing.[citation needed] Electronic sensing of refrigerant gases phased out the use of flame testing for leaks in the 1980s. Similarly, phosgene poisoning is a consideration for people fighting fires that are occurring in the vicinity of freon refrigeration equipment, smoking in the vicinity of a freon leak, or fighting fires using halon or halotron.
The great majority of phosgene is used in the production of isocyanates, the most important being toluene diisocyanate (TDI) and methylene diphenyl diisocyanate (MDI). These two isocyanates are precursors to polyurethanes.
Synthesis of carbonates
Significant amounts are also used in the production of polycarbonates by its reaction with bisphenol A.[4] Polycarbonates are an important class of engineering thermoplastic found, for example, in lenses in eye glasses. Diols react with phosgene to give either linear or cyclic carbonates (R = H, alkyl, aryl):
- HOCR2-X-CR2OH + COCl2 → 1/n [OCR2-X-CR2OC(O)-]n + 2 HCl
Synthesis of isocyanates
The synthesis of isocyanates from amines illustrates the electrophilic character of this reagent and its use in introducing the equivalent of "CO2+":[9]
Such reactions are conducted in the presence of a base such as pyridine that absorbs the hydrogen chloride.
Laboratory uses
In the research laboratory phosgene still finds limited use in organic synthesis. A variety of substitutes have been developed, notably trichloromethyl chloroformate ("diphosgene"), a liquid at room temperature, and bis(trichloromethyl) carbonate ("triphosgene"), a crystalline substance.[10] Aside from the above reactions that are widely practiced industrially, phosgene is also used to produceacid chlorides and carbon dioxide from carboxylic acids:
- RCO2H + COCl2 → RC(O)Cl + HCl + CO2
Such acid chlorides react with amines and alcohols to give, respectively, amides and esters, which are commonly used intermediates. Thionyl chloride is more commonly and more safely employed for this application. A specific application for phosgene is the production of chloroformic esters:
- ROH + COCl2 → ROC(O)Cl + HCl
Although it is somewhat hydrophobic, phosgene reacts with water to release hydrogen chloride and carbon dioxide:
- COCl2 + H2O → CO2 + 2 HCl
Analogously, with ammonia, one obtains urea:
- COCl2 + 4 NH3 → CO(NH2)2 + 2 NH4Cl
Halide exchange with nitrogen trifluoride and aluminium tribromide gives COF2 and COBr2, respectively.[4]
History
Phosgene was synthesized by the British chemist John Davy (1790–1868) in 1812 by exposing a mixture of carbon monoxide and chlorine to sunlight. He named it "phosgene" in reference of the use of light to promote the reaction; from Greek, phos (light) and gene (born).[11] It gradually became important in the chemical industry as the 19th century progressed, particularly in dye manufacturing.
Further information: Use of poison gas in World War I and Second Italo-Abyssinian War
Following the extensive use of phosgene gas in combat during World War I, it was stockpiled by various countries as part of their secret chemical weapons programs.[12][13][14]
In May 1928, eleven tons of phosgene escaped from a war surplus store in central Hamburg.[15] 300 people were poisoned of whom 10 died.[15]
.

US Army phosgene identification poster from World War II
Phosgene was then only frequently used by the Imperial Japanese Army against the Chinese during the Second Sino-Japanese War.[16] Gas weapons, such as phosgene, were produced by Unit 731 and authorized by specific orders given by Hirohito (Emperor Showa) himself, transmitted by the chief of staff of the army. For example, the Emperor authorized the use of toxic gas on 375 separate occasions during the battle of Wuhan from August to October 1938.[17]
Phosgene is an insidious poison as the odor may not be noticed and symptoms may be slow to appear.[18] The odor detection threshold for phosgene is 0.4 ppm, four times the threshold limit value. Its high toxicity arises from the action of the phosgene on the proteins in the pulmonary alveoli, the site of gas exchange: their damage disrupts the blood-air barrier, causing suffocation. It reacts with the amines of the proteins, causing crosslinking by formation of urea-like linkages, in accord with the reactions discussed above. Phosgene detection badges are worn by those at risk of exposure.[4]
Sodium bicarbonate may be used to neutralise liquid spills of phosgene. Gaseous spills may be mitigated with ammonia.[19]
.
TRIPHOSGENE HANDLING
.

Left, reaction vessel with amino acid and triphosgene dissolved in THF; middle, appearance of the reaction mixture after addition of 2,4,6-collidine; and right, appearance of the reaction mixture after microwave irradiation.

Typical glassware standard equipment for the safety phosgenation with phosgene supply from triphosgene: (A) phosgene generator (V = 1 L, T = 85 °C) loaded with 600 g of triphosgene; (B) refluxer (water cooled, T = 15 °C); (C) phosgene line (Viton hose); (D) phosgenation reactor (V = 10 L, T = 110 °C); (E) refluxer (cryostat cooled, T = −30 °C); (F) off-gas line (Viton hose) from the top of the refluxer (E); (G) cooling trap (dry ice cooled, T = −60 °C); (H) off-gas line; (I) cryostat. The assembly of the equipment is somewhat reduced to effect more clarity of the ensemble.
.

.
Phosgene is quantitatively formed from solid triphosgene in a solvent-free and safe process without any reaction heat, catalyzed by planar N-heterocycles with deactivated imino functions.
The rate of phosgene generation is adjustable to the rate of phosgene consumption in the subsequent phosgenation reaction by thermal control, catalyst concentration, and in some cases, specific properties of selected metal phthalocyanines. A thermal runaway reaction of this process is impossible.
.
Use a safer process for generating phosgene.

Decomposition of triphosgene (1a) into carbon tetrachloride, carbon dioxide, and 1 equiv of phosgene (3)
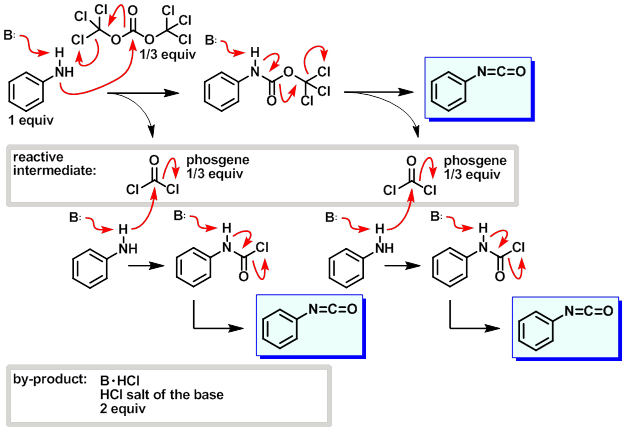
Phosgene (COCl2) is useful in organic synthesis for chlorination, chlorocarbonylation, carbonylation, and dehydration; but its high toxicity discourages its use. Until now, the best substitute for COCl2 has been triphosgene [(CCl3O)2CO], a stable solid that has low vapor pressure. Although (CCl3O)2CO can be used in phosgenation reactions, removing the unreacted reagent from reaction mixtures is difficult because of its high boiling point. In contrast, COCl2 is easily removed by evaporating it.
(CCl3O)2CO reacts with silica gel, metal salts, or Lewis acids to generate 1 equiv of phosgene by an electrocyclic reaction. H. Eckert* and J. Auerweck at the University of Technology, Munich (Germany) report that pyridine and phthalocyanine derivatives catalyze the decomposition of (CCl3O)2CO to generate 3 equiv of COCl2.
The catalysts, phenanthridine , poly(2-vinylpyridine) , and phthalocyanines , convert liquid (CCl3O)2CO to the desired COCl2. The size and structure of the catalysts allow (CCl3O)2CO to react by the mechanism shown. The reaction was run at the 100-g scale to generate 22 L of gaseous COCl2 with an oil bath or an IR heater as the heat source. Because the catalysts are not soluble in (CCl3O)2CO, the process is considered to be heterogeneous catalysis.
.

Controlled transformation of triphosgene (1) into 3 equiv of phosgene (3) catalyzed by 4
Compounds 1 and 4a−4 h are commercially available products from Sigma-Aldrich, with the following purities: 1, 98% (IR νC═O 1820 cm−1, 13C NMR δ 108.0, 140.9); 4a, 98%; 4c, n.a.; 4d, 99%;4e, 97%; 4f, 97%; 4g, 90%; 4h, 85%.
Because the reaction is controlled by temperature, turning off the heat source causes the liquid (CCl3O)2CO to crystallize and stops the reaction, making the process safe. The reaction can be used to generate COCl2 externally or to produce it in situ. According to the authors, this method fulfills the goal of “safety phosgenation on demand of consumer”.
ORGANIC PROCESS RESEARCH AND DEVELOPMENT
Department of Chemistry, Technische Universitaet Muenchen, Lichtenbergstr. 4, Garching 85747, Germany
Org. Process Res. Dev., 2010, 14 (6), pp 1501–1505
DOI: 10.1021/op100239n
READ AT
DETECTION
A FRET approach towards potential detection of phosgene is presented, which is based on a selective chemical reaction between phosgene (or triphosgene as a simulant) and donor and acceptor fluorophores.

.
FRET has been applied in an experimental method for the detection of phosgene. In it, phosgene or rather triphosgene as a safe substitute serves as a linker between an acceptor and a donor coumarine (forming urea groups).[3] The presence of phosgene is detected at 5x10-5M with a typical FRET emission at 464 nm.

Continous Flow
Utilizing a flow-reactor, phosgene precursor can be generated in situwith minimal excess (5%). Since the reaction is done in microliter scale, If the amide is the desired product, immediate amidation, with various amines, will certainly decrease epimerization of the acid chloride. With optimized flow, the reaction can be completed in mere 20 seconds while suppressing generating the other isomer. the results are reproducible. Afterwards, mixture containing the product can be quenched with saturated NH4Cl (aq) in CH2Cl2. Although yield can be slightly lower compared to the batch synthesis, the selectivity is quite strong.
EXAMPLES OF USE OF TRIPHOSGENE
Chlorination of Aliphatic Primary Alcohols via Triphosgene-Triethylamine Activation
Caitlan E. Ayala, Andres Villalpando, Alex L. Nguyen, Gregory T. McCandless and Rendy Kartika*
*Department of Chemistry, 232 Choppin Hall, Louisiana State University, Baton Rouge, Louisiana 70803, United States, Email: rkartika lsu.edu
lsu.edu
Caitlan E. Ayala, Andres Villalpando, Alex L. Nguyen, Gregory T. McCandless and Rendy Kartika*
*Department of Chemistry, 232 Choppin Hall, Louisiana State University, Baton Rouge, Louisiana 70803, United States, Email: rkartika
lsu.edu
C. E. Ayala, A. Villalpando, A. L. Nguyen, G. T. McCandless, R. Kartika, Org. Lett., 2012, 14, 3676-3679.
DOI: 10.1021/ol301520d (free Supporting Information)

Activation of primary aliphatic alcohols with triphosgene and triethylamine mixtures afforded either alkyl chloride or diethylcarbamate products, and the switch in selectivity appeared to be driven by sterics. The reaction conditions to achieve this highly useful transformation were unexceptionally mild and readily tolerated by a wide range of sensitive functionalities.

..............................
.

.
ABACAVIR SULPHATE

VESTIPITANT
The following synthetic route was reported by Giuseppe Guercio et al from GlaxoSmithKline:
The initial chemical development synthetic route, derived from the one used by medicinal chemistry, involved several hazardous reagents, gave low yields and produced high levels of waste. Through a targeted process of research and development, application of novel techniques and extensive route scouting, a new synthetic route for GW597599 was developed. This paper reports the optimisation work of the third and last stage in the chemical synthesis of GW597599 and the development of a pilot-plant-suitable process for the manufacturing of optically pure arylpiperazine derivative 1. In particular, the process eliminated the use of triphosgene in the synthesis of an intermediate carbamoyl chloride, substantially enhancing safety, overall yield, and throughput.

source:
Org. Process Res. Dev., 2009, 13 (6), pp 1100–1110.
Org. Process Res. Dev., 2009, 13 (3), pp 489–493.
Org. Process Res. Dev., 2008, 12 (6), pp 1188–1194.
.
TIVOZANIB

VIAGRA
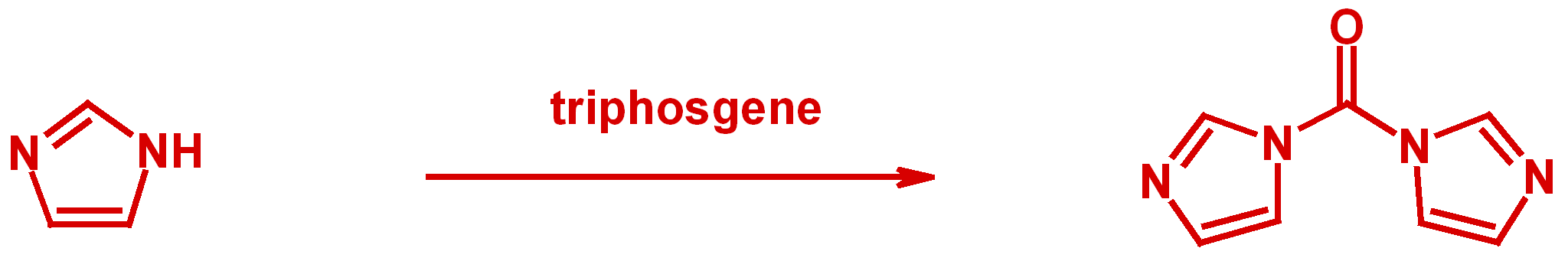
EFAVIRENZ .........EP2454244A1


Enantiomerically pure hydantoins are prepared from optically pure α-amino amides utilizing triphosgene. A mechanism for the racemization observed with 1,1'-carbonyldiimidazole (CDI) for this type of reaction is proposed.
D. Zhang, X. Xing, G. D. Cuny, J. Org. Chem., 2006, 71, 1750-1753.
Double acylation of a titanaselenide by triphosgene;
4,5-ethylenedithio-1,3-diselenol-2-one
N-Hydroxysuccimide esters of carboxylic acids have been widely used in organic synthesis as reactive acylating reagents. These active esters are especially useful as intermediates in the synthesis of peptides and proteins since they acylate primary amines to give the amides in high yields. We have developed a new and convenient one-pot procedure for the preparation of N-hydroxysuccinimide esters of carboxylic acids using N-hydroxysuccinimide and triphosgene as an acid activator. A variety of carboxylic acids can be easily and rapidly converted to the corresponding N-hydroxysuccinimido esters at room temperature. The results of this transformation will be presented.
References
- JMerck Index, 11th Edition, 7310.
- http://www.inchem.org/documents/icsc/icsc/eics0007.htm
- CBRNE - Lung-Damaging Agents, Phosgene May 27, 2009
- Wolfgang Schneider; Werner Diller (2005), "Phosgene", Ullmann's Encyclopedia of Industrial Chemistry, Weinheim: Wiley-VCH, doi:10.1002/14356007.a19_411
- Nakata, M.; Kohata, K.; Fukuyama, T.; Kuchitsu, K. (1980). "Molecular Structure of Phosgene as Studied by Gas Electron Diffraction and Microwave Spectroscopy. The rz Structure and Isotope Effect".Journal of Molecular Spectroscopy 83: 105–117. doi:10.1016/0022-2852(80)90314-8.
- Annex on Implementation and Verification ("Verification Annex")
- https://itportal.decc.gov.uk/cwc_files/S2AAD_guidance.pdf
- "Common Cleaners Can Turn Into Poison Gas". American Iron Magazine. TAM Communications. Retrieved 14 October 2011.
- R. L. Shriner, W. H. Horne, and R. F. B. Cox (1943), "p-Nitrophenyl Isocyanate", Org. Synth.; Coll. Vol. 2: 453
- Hamley, P. "Phosgene" Encyclopedia of Reagents for Organic Synthesis, 2001 John Wiley, New York. doi: 10.1002/047084289X.rp149
- John Davy (1812). "On a Gaseous Compound of Carbonic Oxide and Chlorine". Philosophical Transactions of the Royal Society of London 102: 144–151. doi:10.1098/rstl.1812.0008.JSTOR 107310.
- Base's phantom war reveals its secrets, Lithgow Mercury, 7/08/2008
- Chemical warfare left its legacy, Lithgow Mercury, 9/09/2008
- Chemical bombs sit metres from Lithgow families for 60 years, The Daily Telegraph, September 22, 2008
- Ryan, T.Anthony (1996). Phosgene and Related Carbonyl Halides. Elsevier. pp. 154–155. ISBN 0444824456.
- Yuki Tanaka, "Poison Gas, the Story Japan Would Like to Forget", Bulletin of the Atomic Scientists, October 1988, p. 16–17
- Y. Yoshimi and S. Matsuno, Dokugasusen Kankei Shiryô II, Kaisetsu, Jugonen Sensô Gokuhi Shiryoshu, 1997, p. 27–29
- Borak J., Diller W. F. (2001). "Phosgene exposure: mechanisms of injury and treatment strategies". Journal of Occupational and Environmental Medicine 43 (2): 110–9. doi:10.1097/00043764-200102000-00008. PMID 11227628.
- "Phosgene: Health and Safety Guide". International Programme on Chemical Safety. 1998.
- (a) Cotarca, L. and Eckert, H. Phosgenations − A Handbook; Wiley-VCH: Weinheim, 2003.(b) Cotarca, L. and Eckert, H. Phosgenations − A Handbook; Wiley-VCH:Weinheim, 2003; pp 20− 21.(c) Cotarca, L. and Eckert, H. Phosgenations − A Handbook;Wiley-VCH: Weinheim, 2003; pp 44− 520.(d) Cotarca, L. and Eckert, H. Phosgenations − A Handbook; Wiley-VCH: Weinheim, 2003; p 41.(e) Cotarca, L. and Eckert, H.Phosgenations − A Handbook; Wiley-VCH: Weinheim, 2003; pp 14− 16, 613− 615.
- Recent online information: www.ch.tum.de/oc1/HEckert/research.htm.
- (a) Senet, J. P. The Recent Advance in Phosgene Chemistry; SNPE: Paris, 1997; Vol. 1.(b) Pasquato, L.; Modena, G.; Cotarca, L.; Delogu, P.; Mantovani, S. J. Org. Chem. 2000, 65,8224– 8228(d) Dunlap, K. L. In Kirk-Othmer Encyclopedia of Chemical Technology, 5 ed.;Wiley: New York, 2006; Vol. 18, pp 802− 814.(e) Nielsen, D. H.; Burke, T. G.; Woltz, P. J. H.; Jones, E. A. J. Chem. Phys. 1952, 20, 596– 604(f) Gordon, E. P.;Enakaeva, V. G.; Korotchenko, A. V.; Mitrokhin, A. M. Russian Patent RU 2299852, 2007.
- (a) Eckert, H.; Forster, B. Angew. Chem. 1987, 99, 922– 923 ; Angew. Chem., Int. Ed.,1987, 26, 894–895(b) Eckert, H. TUM-Mitteilungen (Technische Universitaet Muenchen) 2006, 3, 68– 69(d) Triphosgene; Ubichem: U.K., 1999; CD-ROM.
- (a) Eckert, H.; Drefs, N. Chemanager 2006, 3) 10
- Eckert, H.; Dirsch, N.; Gruber, B. (former Dr. Eckert GmbH, now Buss Chem Tech AG) German Offen. DE 19740577, 1999 (Sep. 15, 1997), Chem. Abstr. 1999, 130, 211406.;WO 9914159, 1999; Eur. Pat. EP 1017623, 2002; U.S. Patent US 6399822, 2002; Japanese Patent JP 2001516692, 2001.
- Mole percent 4 referring to 3 phosgene equivalents of 1 .
- (a) Leznoff, C. C.; Lever, A. B. P. Phthalocyanines, Properties and Applications; VCH:Weinheim, NY, 1989.(b) Lever, A. B. P. Adv. Inorg. Chem. Radiochem. 1965, 7, 28– 114(c) Ebert, N. A.; Gottlich, H. B. J. Am. Chem. Soc. 1952, 74, 2806[ACS Full Text
 ], [CAS]
], [CAS] - The weighing error of this procedure mainly comes from icy condensed humidity at the cool glassware of the cooling trap and is less than 0.5 g, determined by a series of weighings under the same conditions, the same equipment, temperature (T = −78 °C), and handling time <10 s, but without 3. Under these conditions evaporation of 3 (bp 8 °C) hardly ever happens and can be ignored.
- Monitox plus gas monitor (COCl2) and phosgene badges from Compurhttp://www.compur.com/gasmessgeraete/front_content.php?idcat=7&changelang=3.
- Davy's account of his discovery of phosgene
- International Chemical Safety Card 0007
- CDC - Phosgene - NIOSH Workplace Safety and Health Topic
- NIOSH Pocket Guide to Chemical Hazards
- U.S. CDC Emergency Preparedness & Response
- U.S. EPA Acute Exposure Guideline Levels
- Regime For Schedule 3 Chemicals And Facilities Related To Such Chemicals, OPCW website
- CBWInfo website
- Use of Phosgene in WWII and in modern-day warfare (Refer to Section 4.C of the article)
- An experience with accidental poisoning by heated tetrachlorethylene solvent
Tetramethylpyrazine could protect against CoCl2 -induced neurotoxicity in PC12 cells and in rats, as evidenced by enhancement of cell viability in PC12 cells and improvement of learning and memory ability in rats treated by CoCl2. Tetramethylpyrazine
ReplyDeleteThat is a really excellent piece anyone needs to investigate thank you for sharing. When using a ffree auto clicker, it's recommended to ensure the software is reputable, comes from a trusted source, and is used within legal and ethical boundaries. I hope you also visit my blog and give us a good opinion.
ReplyDelete